

Insuficiencia suprarrenal primaria por adrenalitis autoimnume
La insuficiencia suprarrenal puede ser primaria (suprarrenal), secundaria (hipofisiaria) o terciaria (hipotalámica); a su vez puede ser completa (afectando a los 3 esteroides suprarrenales) o parcial.
Santiago Muzzo B (1,2), Gianina Izquierdo C. (2), Sandra Verbeke P (3,4) Rev. Méd. Chile, agosto 2002. V.130, N.8, Santiago.
1 Unidad de Endocrinología del INTA, Universidad de Chile
2 Servicio de Pediatría, Clínica Alemana de Santiago, Chile
3 Unidad de Gastroenterología del INTA,Universidad de Chile
4 Laboratorio de Clínica Santa María,Santiago, Chile
Correspondencia: smuzzo@uec.inta.uchile.cl
La insuficiencia suprarrenal puede ser primaria (suprarrenal), secundaria (hipofisiaria) o terciaria (hipotalámica); a su vez puede ser completa (afectando a los 3 esteroides suprarrenales) o parcial. Se puede sospechar por el cuadro clínico y se confirma por el déficit de cortisol plasmático y de mineralocorticoides. Se puede producir por destrucción del tejido (traumáticas o tumores), en otros casos puede ser congénita y comprobarse por exámenes específicos. Actualmente la causa más frecuente de insuficiencia suprarrenal es la autoinmune (Tabla 1).

Es de interés la presentación de este caso clínico de insuficiencia suprarrenal crónica secundaria a adrenalitis autoinmune aislada, por la importancia del diagnóstico temprano, por sus características inmunológicas y por la excelente respuesta al tratamiento. Los antecedentes clínicos del paciente descartan la mayoría de las etiologías de insuficiencia suprarrenal, motivo por el que sólo se analizarán la adrenoleucodistrofia y la adrenalitis autoinmune en el diagnóstico diferencial.
Caso Clínico
CVR, escolar de 10 años de edad, de sexo masculino, que presentaba un rendimiento escolar adecuado y una actividad física frecuente hasta 3 meses antes de su ingreso, cuando inició un cuadro de astenia, anorexia con pérdida de 4 kg de peso corporal y adinamia progresiva e incapacitante hasta quedar postrado.
Fue producto de un primer embarazo de padres jóvenes, sanos y de un parto normal. Presentó ictericia neonatal tratada con fototerapia.
Entre sus enfermedades anteriores destacaba una constipación crónica secundaria a un dolicomegacolon y un reflujo gastro-esofágico leve.
A los síntomas iniciales se agregaron náuseas y vómitos intensos, motivo por el que se hospitalizó en Viña del Mar con los diagnósticos de deshidratación, acidosis metabólica severa y astenia. Al ingreso presentaba un pH en sangre de 7,2; HCO3 de 12 mmol/L; BE: -12,6 mmol/L, natremia 124 mEq/L, potasemia 4,9 mEq/L y creatininemia 2,4 que bajó posteriormente a 0,68 mg/dL. A pesar de la infusión intravenosa de solución salina y bicarbonato, continuó en acidosis metabólica, natremias bajas y potasemias en el rango normal-alto.
Evaluado por nefrólogo, gastroenterólogo y neurólogo, quienes solicitaron: examen de orina, uremia, aminoaciduria, aminoacidemia, anticuerpos antinucleares, radiografías de cráneo y huesos largos, ecografía de abdomen, TAC cerebral y cintigrafía ósea que resultaron normales. Después de 13 días de hospitalización fue dado de alta sin diagnóstico, reingresando 3 días después con mayor compromiso del estado general, deshidratación severa, somnolencia, debilidad extrema que dificultaba la marcha, acidosis metabólica y natremia de 98 mEq/L que se corregía sólo hasta 112 mEq/L con solución salina endovenosa.
Se trasladó a la UCI de Clínica Alemana, ingresando afebril, con taquicardia (105 x min), una presión arterial de 105/55 mm de Hg, diuresis de 3,3 cc/kg/h. Muy decaído, ligeramente obnubilado, con enlentecimiento de sus respuestas. Enflaquecido, hidratado, sin melanoplaquias bucales y con disminución simétrica de la fuerza muscular en las extremidades inferiores.
El laboratorio reveló un hematocrito de 32%, hemoglobina 11,2 g/dL, 5.400 leucocitos por mm3 sin desviación a izquierda, 53,7% de neutrófilos y 36,6% de linfocitos; VHS 10 mm/hr, PCR 0,49 mg/dL, natremia 116 mEq/L,potasemia 4,3 mEq/L, cloremia 93 mEq/L; pH 7,4; pCO de 27,3 mmHg, HCO : 17,7 mmol/L y 2 3 BE de -4,9 mmol/L; la osmolalidad plasmática 245 mOsm/Kg H O, la urinaria de 24 h 258 2 mOsm/Kg H O (VN 275-295); sodio en orina: 59 2 mEq/L, creatinina 0,27 mg/dL, glicemia 100 mg/dL, perfil bioquímico con un nitrógeno ureico 1,4 mg/dL, uremia 3,0 mg/dL(VN 3,8- 5,4), uricemia 2,0 mg/dL (VN 2,2-6,6), albúmina 3,4 g/dL, transaminasas normales, triglicéridos: 45 mg/dL (30-100). La resonancia nuclear magnética (RNM) cerebral mostró una alteración de la sustancia blanca compatible con alteraciones hidroelectrolíticas, el electrocardiograma (ECG) detectó taquicardia sinusal y ecocardiograma normal, la radiografía (Rx) de tórax informó microcardia. Se trató el trastorno electrolítico con una solución de NaCl al 3%, la mitad en las primeras 8 h y el resto en las 16 h siguientes, subiendo la natremia hasta 125 mEq/L. A las 24 h la natremia fue 123 mEq/L, potasemia 4,39 mEq/L y cloremia 93 mEq/L, la osmolalidad plasmática 258 mOsm /KgH2O.
A las 24 h del ingreso se detectó un cortisol plasmático PM de 3,39 ug/dL (VN: 3-16), el ritmo circadiano de cortisol fue 3,24 ug/dL AM (VN: 3-51) y de 3,91 ug/dL PM (16-17 h) (VN: 3 a 16), el cortisol libre en orina 1,7 ug/24 h (VN: 2 a 27) y la prueba de estimulación con 1 mg de Synacthen, plano, confirmándose una insuficiencia suprarrenal primaria.
Se completó el estudio con T3: 1,17 ng/mL (VN 0,8-2,6), T4: 9,78 ug/dL (VN 6-13), TSH: 2,13 uU/mL (VN 0,7 – 6,4) , anticuerpos antitiroglobulina y antimicrosomales negativos, testosterona total 0,29 ng/mL, actividad de renina plasmática 43,7 ng/mL/h (VN: 0,5 a 3,3) y aldosterona plasmática
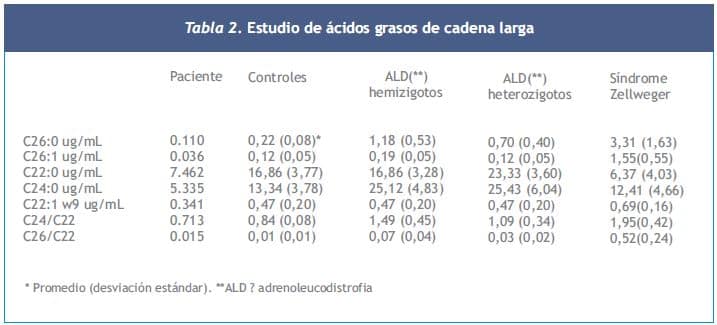
Los ácidos grasos de cadena larga se analizaron por cromatografía capilar de gas en el Kennedy Krieger Institute de Baltimore, USA, fueron normales (Tabla 2).
Con el diagnóstico de insuficiencia suprarrenal crónica primaria, secundaria a adrenalitis autoinmune se inició el tratamiento con una dosis de carga de 50 mg de hidrocortisona, luego cortisona oral 25 mg cada 12 h y Florinef (9 alfa fluor hidrocortisona) de 100 ug, 1/2 tableta cada 12 h, evolucionando en forma muy favorable, con aumento y normalización progresiva de la natremia y de la cloremia, manteniendo la presión arterial en un rango entre 79/45 y 103/51 mmHg, con taquicardia compensadora (110 a 130 x min). El laboratorio detectó una hemoglobina de 8,3 g/dL tratándose con sulfato ferroso. Al tercer día de hospitalización se trasladó a sala y se inició la realimentación oral.
Se detectó una dificultad en la marcha con hiperestesia en la región plantar y de los ortejos. Evaluado por neurólogo observó una hipotrofia muscular generalizada, con franca disminución simétrica de ROT en extremidades inferiores, siendo catalogado como neuritis periférica secundaria al trastorno metabólico que presentó, que fue regresando espontáneamente. Fue dado de alta en buenas condiciones generales, con un tratamiento sustitutivo de cortisona (20 mg) 0,5 tableta cada 12 h y Florinef (100 ug) 1 tableta al día. Los controles ambulatorios han demostrado una normalización total de su estado de salud.
Discusión
El diagnóstico no es fácil para el pediatra, que habitualmente no piensa en esta enfermedad dada su baja frecuencia. Debería sospecharse en un paciente que presenta cuadros de deshidratación no bien explicados, con astenia y adinamia progresiva, pigmentación de mucosas y con un laboratorio con hiponatremia marcada, especialmente si se acompaña de hiperpotasemia (1,2).
Muchas de las etiologías de la insuficiencia suprarrenal se pueden descartar con una buena historia clínica, como en la hemorragia suprarrenal
del recién nacido, secundarias a traumatismos, producidas por enfermedades infecciosas como TBC, meningococcemia y parasitosis.
Algunos exámenes de laboratorio específicos pueden descartar otras etiologías como la 17 OH progesterona plasmática en el caso de la hiperplasia suprarrenal congénita. Es interesante destacar la rápida recuperación que pueden tener estos pacientes, que suelen llegar en condiciones muy graves.
Una insuficiencia suprarrenal acompañada de deterioro neurológico progresivo previo o posterior a la presentación de este síndrome, debe hacer pensar en una adrenoleucodistrofia (ALD-X), enfermedad que se diagnostica midien-do los ácidos grasos de cadena muy larga (AGCML). Es una afección genética peroxisomal ligada al cromosoma X, caracterizada por un compromiso neurológico progresivo (leucodistrofia) y adrenal (insuficiencia suprarrenal) producto de la acumulación de AGCML (1). La ausencia de la proteína ALDP necesariapara el transporte de los AGCML, sería la responsable de la acumulación de AGCML propia de esta afección (3,9).
En el caso clínico presentado se descartaría esta enfermedad dado que el paciente no presentaba signos neurológicos y la determinación de los AGCML efectuada fue normal.
La etiología autoinmune es la más frecuente y puede darse en forma aislada o como parte de un síndrome poliglandular autoinmune. La adrenalitis autoinmune (AA) puede ser familiar o no, siendo menos frecuente que tenga agrupación familiar cuando se presenta en forma aislada (30%), a diferencia de los síndromes poliglandulares los cuales en 50% presentan compromiso familiar.

El descubrimiento de autoanticuerpos contra las células adrenales indicó su origen autoinmune, siendo de mucha utilidad en el diagnóstico diferencial de la insuficiencia suprarrenal (10,11). Los anticuerpos antiadrenales se detectaron por inmunofluorescencia indirecta (IFI) al exponer suero de mujeres con enfermedad de Addison con tejido adrenal, ovario, testículo y placenta humana y de mono. Los anticuerpos reaccionan con el citoplasma de las células de diferentes zonas de la glándula adrenal y de células esteroidales de otros tejidos, indicando que los antígenos blancos están presentes en las células productoras de esteroides de las diferentes glándulas involucradas en las manifestaciones clínicas (12,13). El antígeno blanco principal es un citocromo p450, responsable de la producción de testosterona, estrógenos, cortisol y aldosterona (14). La presencia de estos anticuerpos indica adrenalitis y ooforitis autoinmune, si bien su rol patogénico no está claro, ya que los anticuerpos podrían ser consecuencia de la destrucción de las células endocrinas más que su causa (13,15).
La Tabla 3 muestra la prevalencia de estos anticuerpos. En el síndrome poliglandular autoinmune tipo 1 (SPGA1) tienen un valor predictivo del desarrollo de la enfermedad en 92% de los casos. Su presencia es indicativa de riesgo de insuficiencia suprarrenal en niños más que adultos y su título se correlaciona con el grado de disfunción suprarrenal (13,15,17).

La AA puede ser parte del síndrome poliglandular autoinmune tipo 1 o tipo 2 o presentarse en forma aislada. La mitad de los pacientes con insuficiencia suprarrenal autoinmune tienen al menos otra glándula endocrina comprometida.
Ser parte del síndrome poliglandular es más frecuente en el sexo femenino (70%), en cambio los casos aislados son más frecuentes en varones (71%) (18).
El paciente con SPGA 1 desarrolla candidiasis mucocutánea crónica poco después de nacer y luego hipoparatiroidismo autoinmune y enfermedad de Addison. Puede tener otras alteraciones como gastritis autoinmune con o sin anemia perniciosa, hepatitis autoinmune, diabetes tipo 1, enfermedad tiroidea autoinmune, alopecía, vitiligo, queratopatía, enfermedad celíaca e hipogonadismo (16) (Tabla 4).
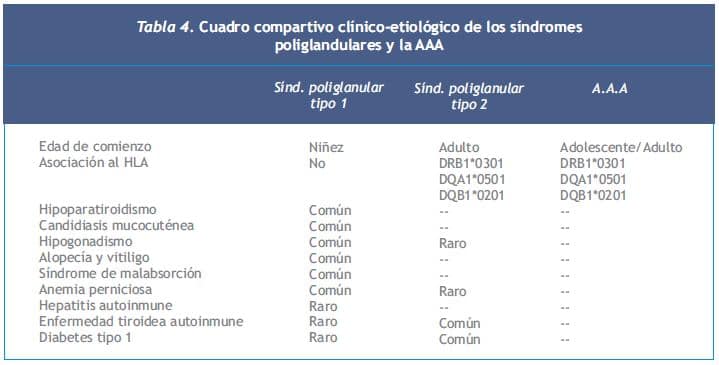
En el paciente con síndrome poliglandular autoinmune tipo 2 (SPGA 2) la insuficiencia suprarrenal es su principal manifestación. Son frecuentes la tiroiditis autoinmune crónica y la diabetes tipo 1. En el 50% de los casos la manifestación inicial es el compromiso adrenal, ocurriendo simultáneamente con una enfermedad tiroidea autoinmune o diabetes tipo 1 en 20% de los casos (17).

La AA aislada (AAA) se presenta a la misma edad de comienzo que el SPGA 2 y se asocia al HLA. No se observan otros desórdenes endocrinos, considerándose una variante del SPGA 2 por la asociación con el HLA y la presencia de anticuerpos antitiroideos, gástricos y células de los islotes (Tabla 4). Los anticuerpos plasmáticos están presentes en 60 a 75% de los pacientes con insuficiencia suprarrenal primaria, son más frecuentes en mujeres, principalmente aquellas con síndromes poliglandulares. 19% de los pacientes con anticuerpos (+) desarrollan insuficiencia suprarrenal dentro de un año.
El diagnóstico de la insuficiencia suprarrenal debe sospecharse precozmente para evitar el riesgo de vida de estos pacientes al retrasar el inicio de su tratamiento. Es importante que el pediatra general y los intensivistas tengan en cuenta esta enfermedad, que a pesar de su baja frecuencia tiene un excelente resultado terapéutico.
Referencias Bibliográficas
1. Raymonde F, Gagnon, Halperin ML. Possible mechanisms to explain the abscence of hiperkalaemia in Addison’s disease. Nephrol Dial Transplant 2001; 16: 1280-4.
2. Barnett AH, Espiner EA, Donald RA. Patients presenting with Addison’s disease need not been pigmented. Postgrad Med J 1982; 58: 690-5.
3. Mosser J, Duoar AM, Sarde CO et al. Putative X-linked adrenoleucodistrophy gene shares unexpected homology with ABC transporters. Nature 1993; 361: 726-30.
4. Bezman L, Moser HW. The incidence of X-linked adrenoleucodistrophy and the relative frecuency of its phenotypes. Am J Med Genet 1998; 76: 415-9.
5. Migeon BR, Moser HW, Moser AB, Axelman J, Sillence D, Norum RA. Adrenoleucodistrophy: Evidence for Xlinkage, inactivation and selection favoring the mutant allele in heterozygous cells. Proc Natl Acad Sci USA 1981; 78: 5066-70.
6. Igarashi M, Achaumburg HH, Powers J, Kishimoto Y et al. Fatty acid abnormality in adrenoleucodistrophy. J Neurochem 1976; 26: 851-60.
7. Moser HW, Moser AB, Smith KD, Bergin A et al. Adrenoleucodistrophy: Phenotypic variability: Implications for therapy. J Inher Metab Dis 1992; 15: 645- 64.
8. Moser HW, Moser AB, Frayer KK et al. Adrenoleucodistrophy:increased plasma content of saturated very long chain fatty acids. Neurology 1981; 31: 1241-9.
9. Moser HW. Adrenoleucodistrophy: Phenotype, genetics, pathogenesis and therapy. Brain 1997; 120: 1485-508.
10. Peterson P, Uibo R, Krohn JE. Addison disease. J Clin Endocrinol Metab 1997; 82: 932-8.
11. Sotssiou F, Bottazzo GF, Doniach D. Immunofluorescence on autoantibodies to steroidproducing cells and to germ line cell in endocrine disease and infertility. Clin Exp Immunol 1980; 39: 97-111.
12. Hoek A, Wulffrat NM, Drexhage HA. Steroid cell autoantibodies. In: Steroid cells autoantibodies. Eds.:
Peter JB, Shonenfeld Y. Elsevier, Amsterdam, The Netherlands 1996; 798-805.
13. Riley W. Enzymes as antigens in autoimmune endocrinopathies. Clin Chem 1995; 41: 337-9.
14. Betterle C, Rossi A, Dalla Priat S, Artifoni A, Pedini B, Gavaso S, Caretto A. Premature ovarian failure: autoimmunity and natural history. Clin Endocrinol 1993;39: 35-43.
15. Betterle C, Volpato M et al. Adrenal cortex and steroid 21-hydroxylase autoantibodies in children with organspecific autoinmune diseases: markers of high progression to clinical Addison disease. J Clin Endocrinol Metab 1997; 82: 939-42.
16. Betterle C, Greggio NA, Volpato M. Autoimmune polyglandular syndrome type I. J Clin Endocrinol Metab 1998; 83: 1049-55.
17. Ahonen P et al. Clinical variation of autoimmune polyendocrinopathy-candidiasis-ectodermal distrophy (APECED) in a series of 68 patients. New Engl J Med 1990; 322: 1829-36.
18. Betterle C, Scalisi C, Presotto F et al. The natural history of adrenal function in autoimmune patients with adrenal autoantibodies. J Endocrinol 1998; 117: 467-71.

Más notas de la edición 6
Lee nuestra última edición publicada en Marzo 2026, Edición número 175

Notas relacionadas a Insuficiencia suprarrenal primaria por...


