



Variedades de debut temprano en la Enfermedad de Pompe
Myriam Ley Martos. Unidad de Gestión Clínica de Pediatría. Hospital Universitario Puerta del Mar Cádiz.
Asociación Española de Enfermos de Glucogenosis. Javier Fernandez Salido, Alberto Molares Vila, Jesus Sueiro Justel, Juan J. Albero Samper, Benjamín Anton, Antonio M. Banon Hernandez, Laura Castells Molines, Jose Luis Ceide Arias, Leonor Fernandez Marcos, Maria Jose Santos García
1.- Introducción
La enfermedad de Pompe (OMIM 131300) [1] es una enfermedad metabólica hereditaria extremadamente rara, incluida dentro de los errores innatos del metabolismo, que pertenece al grupo de las glucogenosis.
Las glucogenosis son enfermedades de depósito lisosomal que afectan a la formación y a la utilización del glucógeno, originando concentraciones o estructuras anormales del mismo [2].
Su origen es genético y se manifiesta con un espectro continuo de síntomas que van aumentando con el paso del tiempo.
Está causada por un déficit de la enzima lisosomal alfa glucosidasa ácida (GAA) también llamada Maltasa ácida.
Su frecuencia se estima entre 1/40.000 a 1/150.000 nacidos vivos. No hay preferencia en cuanto al sexo o el origen étnico. Aún se está estudiando el perfil clínico por lo que es muy útil participar en el registro internacional de la enfermedad (www.pomperegistry.com).
2.-Etiología
La enfermedad de Pompe produce la acumulación progresiva de glucógeno en prácticamente todas las células del organismo, aunque las alteraciones más evidentes se producen en el corazón, el músculo esquelético y en el sistema nervioso.
Su herencia sigue un patrón autosómico recesivo. El gen afectado se encuentra en el brazolargo del cromosoma 17 (locus 17q25) y tiene 19 exones codificantes. En la actualidad, hay descritas más de 340 mutaciones. La escasa frecuencia de la enfermedad dificulta su estudio, tanto desde el punto de vista clínico como en el de la investigación, lo que ha llevado a formar un registro internacional. Este registro permite conocer las diferentes mutaciones que se relacionan con la enfermedad y la correlación con la gravedad clínica de la misma.
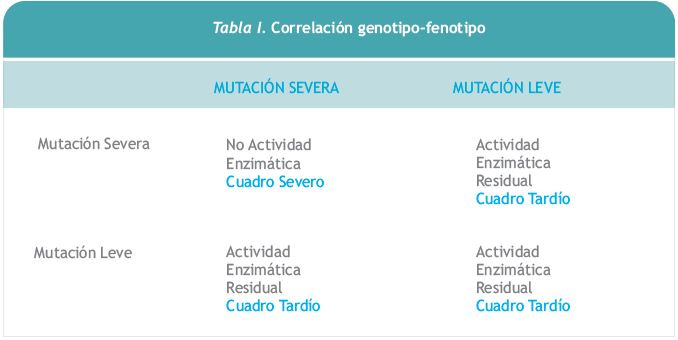
No todas las mutaciones son del mismo tipo. Se han descrito mutaciones missense, nonsense, defectos en splicing, deleciones e inserciones. Para que la enfermedad se manifieste tienen que estar afectados los 2 alelos [3]. Algunos trabajos indican que el curso clínico de la enfermedad se establece principalmente por la naturaleza de la mutación en los dos alelos lo que ocasiona diferentes grados de deficiencia enzimática [4,5]. De esta forma, si las dos mutaciones son severas originará una clínica más grave y los síntomas aparecerán más precozmente en el tiempo debido a la falta total de actividad enzimática. Si las mutaciones son menos agresivas puede haber actividad enzimática aunque deficiente, con lo que la clínica será menos agresiva y la progresión será más lenta [6]. La enfermedad se desarrolla cuando la actividad de GAA es menor del 30% de la considerada normal. (Tabla I y II). No obstante, la correlación genotipo-fenotipo no es total ya que también intervienen factores moduladores genéticos y epigenéticos [7].

3.- Variedades Clínicas
Clásicamente se han descrito dos formas clínicas según la edad de comienzo de los síntomas:
- Forma infantil precoz: cuando la clínica aparece antes del año de vida.
- Forma de inicio tardío: cuando los síntomas aparecen más tarde.
El momento de aparición y la gravedad en la progresión de la enfermedad depende del porcentaje de actividad enzimática, siendo las formas precoces las que tienen menor actividad y por tanto son las de peor pronóstico.
En realidad se produce un espectro continuo de síntomas que va desde la mayor posible, pudiendo aparecer incluso en periodo neonatal con actividad enzimática cero, hasta una forma más leve que se inicia en el adulto, ocasionando solamente debilidad de la musculatura respiratoria y de los miembros [8]. Esta forma del adulto puede tener una progresión estable durante años siendo difícil establecer el momento exacto de aparición de los síntomas.
Forma infantil precoz
Los síntomas aparecen antes del año de edad. Dentro de ella pueden verse dos tipos:
- Forma clásica del lactante, de inicio en los primeros meses y de extrema gravedad, llevando al paciente al fallecimiento en los 2 primeros años de la vida.
- Forma no clásica o atípica del lactante, de menor gravedad y evolución más lenta.
Forma infantil tardía
Con aparición de los síntomas después del año, de las que se conocen dos variantes clínicas:
- Forma infantil o juvenil
- Forma del adulto
En las formas de curso lento es muy difícil definir si los síntomas empezaron antes o después del año ya que estos no aparecen hasta que la deficiencia de la función del órgano llega a un determinado nivel.
4.- Síntomas
En el recién nacido el síntoma más llamativo es la hipotonía y la debilidad. La movilidad espontánea y la refleja están disminuidas y son frecuentes los problemas con la alimentación por succión débil. Ello provoca escasa ganancia de peso. La debilidad a nivel del diafragma origina procesos respiratorios frecuentes, con tos débil y poco eficaz. La hipotonía, especialmente a nivel del tronco hace que tengan mal control cervical, con dificultad para mantener la cabeza erguida. Los reflejos musculares profundos están muy disminuidos o abolidos. Clínicamente se manifiesta como un retraso en el desarrollo motor puro, estando preservadas otras áreas del desarrollo, como el contacto afectivo y la sonrisa, la persecución ocular y el balbuceo.
En la forma clásica del lactante suele asociarse cardiomegalia antes de los 6 meses de vida generalmente por hipertrofia del tabique interventricular, por lo que son frecuentes encontrar síntomas cardiológicos asociados, como insuficiencia cardiaca congestiva.
Más de la mitad asocian macroglosia y hepatomegalia con elevación de las enzimas hepáticas y musculares. Pueden estar presentes además reflujo gastroesofágico y apneas de sueño.
En la forma atípica del lactante. Los síntomas son de inicio muy precoz pero de evolución lenta, sin miocardiopatía hipertrófica o muy leve afectación y sobreviven generalmente a los 2 años.

La enfermedad de comienzo tardío puede manifestarse en cualquier momento después del año, con clínica lentamente progresiva. (Tabla III). Existen síntomas y signos guía que pueden orientar la clínica para el diagnóstico de la enfermedad de Pompe de inicio precoz clásica.

Estos signos son:
- Debut a los 2 o 3 meses
- Poca movilidad
- Dificultad para la succión y deglución
- Escasa ganancia de peso
- Retraso exclusivamente motor con preservación de otras áreas de desarrollo
- Cardiomegalia (92%)
- Elevación de transaminasas y enzimas musculares (GOT, GPT, LDH y aldolasa)
- Infecciones pulmonares repetidas
- Macroglosia (50%)
En la variante tardía, el motivo de consulta suele ser la debilidad y el escaso desarrollo muscular, la hipotonía y a veces las mialgias. Es también frecuente la dificultad para subir escaleras, la marcha anormal, las desviaciones de la columna, las escápulas aladas, la fatiga con el ejercicio, las apneas de sueño, las cefaleas matutinas o la somnolencia diurna por hipo ventilación durante el sueño.
Estos síntomas son compartidos con otras enfermedades musculares. Este hecho unido a la baja frecuencia de la enfermedad y la progresión lentamente progresiva hace que el diagnóstico definitivo pueda retrasarse en el tiempo.
5.- Diagnóstico
Análisis de laboratorio
La primera aproximación diagnóstica en un lactante con hipotonía es la determinación analítica, con transaminasas y enzimas musculares (CPK, GOT, GPT, Aldolasa) que suelen estar elevadas.

Electromiografía-Electroneurografía
El estudio electromiográfico-neurográfico (EMG-ENG) es de difícil interpretación en los lactantes pequeños. Puede ser normal al inicio o bien orientar a patología periférica de origen muscular (patrón miopático) con normalidad en el patrón neurógeno. Es característico aunque no frecuente la aparición de descargas pseudomiotónicas repetitivas. Esta característica, cuando aparece, puede ser muy valiosa para hacer el diagnóstico diferencial ya que no aparecen en otras enfermedades con participación muscular de inicio precoz. Puede apreciarse además potencial de fibrilación y excesiva irritabilidad eléctrica.
Estudio cardiológico
Como en cualquier sospecha de enfermedad de probable origen muscular es necesario hacer un estudio cardiológico aunque aún se encuentre asintomático, que incluya radiografía de torax, electrocardiograma (ECG) y ecocardiografía. El ECG en la enfermedad de Pompe muestra acortamiento del espacio P-R y complejos QRS gigantes [10 y 11].
Screening en papel de filtro (gota seca)
El estudio en gota seca es un método de screening. En algunos países se esta utilizando para detectar pacientes afectados por la enfermedad en periodo pre-sintomático. También puede ser útil como primer paso en la orientación diagnóstica de un paciente con una clínica sugestiva. El estudio se hace con gotas de sangre seca en papel de filtro con técnicas fluorimétricas. Es una técnica de bajo costo y sobre todo rápida.
Screening por detección de tetrasacárido de glucosa (Glc4)
Se ha desarrollado un test de Screening para el lactante que sirve además para monitorizar la respuesta al tratamiento enzimático sustitutivo basado en la detección en orina del tetrasacárido de glucosa (Glc4). Este sacárido está muy aumentado en los pacientes con enfermedad de Pompe y disminuye en los tratados con la enzima [12 y 13].
Determinación de la actividad enzimática de alfa glucosidasa ácida
En caso de ser positivo, la confirmación diagnóstica debe establecerse con el estudio de la actividad enzimática de la alfa glucosidasa ácida en linfocitos sanguíneos o fibroblastos procedentes de biopsia de piel. Esta última técnica es lenta, ya que los fibroblastos tienen que ser cultivados y no es conveniente demorar el diagnóstico ya que la precocidad en la instauración del tratamiento es fundamental en el pronóstico de la enfermedad.

Biopsia muscular
La biopsia muscular ha sido considerada hasta hace pocos años la prueba diagnóstica definitiva. Actualmente, se considera que no es siempre necesaria si el diagnóstico está claro en la forma infantil precoz. Si puede ser necesaria en las formas de inicio tardío aunque no es infrecuente que el resultado sea normal. En el análisis microscópico de la biopsia se pueden apreciar los depósitos vacuolares de glucógeno en las fibras musculares. En adultos con sospecha diagnóstica y biopsia muscular normal puede recurrirse al estudio de la actividad enzimática.
Estudio genético de las mutaciones
Completa el estudio la investigación de las mutaciones génicas, debiendo ser portadores de una mutación cada uno de los progenitores. El estudio genético es fundamental al inicio del tratamiento (como confirmación diagnóstica), para el estudio de portadores y para el diagnóstico prenatal.
Síntomas y signos guía
La hipotonía en el lactante es un signo neurológico frecuente e inespecífico que puede encontrarse en numerosas enfermedades. Para el pediatra, el hecho de la muy baja frecuencia de la enfermedad de Pompe de inicio precoz puede hacer difícil que piense en un primer momento en la enfermedad cuando detecta hipotonía en un lactante. En estos casos la confluencia en el paciente de varias características puede facilitar la orientación diagnóstica.
Estas características son:
- Normalidad al nacer
- Debut temprano de los síntomas (2-4 meses)
- Ausencia de rasgos dismórficos
- Retraso motor puro con debilidad proximal
- No hipoglucemia
- Macroglosia progresiva
- Enfermedad muscular con afectación cardiaca muy precoz, especialmente
- Miocardiopatía hipertrófica y alteraciones de la conducción, ECG con complejos QRS grandes, PR corto y trastornos de repolarización
- EMG con patrón miopático y descargas pseudomiotónicas
- Afectación de enzimas hepáticas con bilirrubina normal
- Proporción entre transaminasas y CPK:(CPK/GOT: 1/2; CPK/GPT: 1/5).
EMG: Electromiograma
ECG: Electrocardiograma
CPK: Creatin fosfoquinasa
GPT: Transaminasa glutámico pirúvica
GOT: Transaminasa glutámico oxalacética

Diagnóstico prenatal
Se recomienda que las familias con antecedentes de la enfermedad, especialmente la forma infantil precoz realicen diagnóstico prenatal en posteriores gestaciones. Puede realizarse midiendo la actividad enzimática a partir de amniocentesis, generalmente entre las semanas 15 y 16 de gestación, o por medición de la actividad enzimática por biopsia de vellosidades coriónicas, usando 4MUG como substrato, entre las semanas 10 y 11 de gestación.
6.- Diagnóstico diferencial
Diversas enfermedades pueden presentar una clínica parecida, teniendo en cuenta que uno de los síntomas más precoces es la hipotonía, signo muy frecuente pero poco específico.
Los principales trastornos que deben barajarse dependen de la forma de inicio.
Forma infantil precoz
El diagnóstico diferencial debe hacerse principalmente con:
- Atrofia muscular espinal tipo I (AME I): Afectación neurógena en el EMG-ENG
- Fibroelastosis endocárdica: Miocardiopatía más acusada en cavidades izquierdas
- Miocarditis
- Distrofia muscular congénita
- Miopatías congénitas
- Enfermedades peroxisomales
- Enfermedades mitocondriales: Frecuente oftalmoplejía. Algunas con crisis epilépticas
- Hipotiroidismo: perfil tiroideo anormal
- Miocardiopatía hipertrófica idiopática: Hipertrofia biventricular. No afectación músculo esquelético
Forma de comienzo tardío
Las enfermedades que se precisa diferenciar de la enfermedad de Pompe son:

- Distrofinopatías (Duchenne o Becker)
- Dermatomiositis, polimiositis
- Distrofia muscular de cinturas
- Síndrome escápulo perineal
- Distrofia muscular congénita
- Enfermedad de Danon (Tipo IIb). Gran cardiomiopatía hipertrófica, poca afectación muscular, retraso mental. Actividad AGG normal. Sediferencian por biopsia muscular. La mayoría delas veces el diagnóstico precisa de la biopsiamuscular.
7.- Tratamiento
En la actualidad no existe tratamiento curativo de la enfermedad. La terapia enzimática sustitutiva (TES) es una opción terapéutica actualmente disponible. Esta terapia si bien no es definitiva, si enlentece la progresión de la enfermedad y mejora los síntomas en las formas infantiles precoces si se comienza antes del deterioro clínico. Esta circunstancia hace que la precocidad en el diagnóstico sea un elemento clave para mejorar el pronóstico y alargar la esperanza de vida ganando tiempo en espera de un tratamiento definitivo que actualmente están en fase de investigación.
Tratamiento de mantenimiento
- Terapia enzimática sustitutiva: Alglucosidasaalfa (alfa 1,4 glucosidasa humana recombinante R Myozyme), intravenosa a una dosis media de 20 mg/Kg que se administra cada 2 semanas. Se recomienda un ritmo de infusión lento de aproximadamente 1 mg/Kg/hora con incremento gradual de 2 mg/Kg cada 30 minutos hasta llegar a 7 mg/Kg/hora. Para evitar reacciones adversas puede premedicarse con antitérmicos y antihistamínicos (cuando la edad del paciente lo permita). Cuando los pacientes adquieren un peso adecuado, para facilitar el acceso venoso, se les coloca un sistema tipo Port-a-cath® implantado generalmente en la vena subclavia.
La enzima es mejor tolerada en los pacientes que tienen actividad enzimática a la creación de anticuerpos tipo IgG neutralizantes. Se está investigando la posibilidad de inmunomodulación, reducción o incluso inhibición de la respuesta inmunológica producida por la administración de la enzima [14 y 15]. Indicado en las formas infantiles precoces antes del deterioro. En las formas tardías se recomienda individualizar cada caso.
Son factores de mal pronóstico en cuanto a respuesta favorable al tratamiento:
- Ausencia de actividad enzimática residual (CRIM -)
- Determinados genotipos: stop codons, null mutation
- Biopsia muscular con extenso daño y numerosas vacuolas en músculo
- Comorbilidad, gastrostomía, traqueotomía
- Severas alteraciones de la deglución
- Shock cardiocirculatorio
Se recomienda discontinuar el tratamiento en los siguientes casos:
- Shock anafiláctico (posibilidad de desensibilización, con dilución).
- Escasa respuesta al tratamiento, valorado con escala de calidad de vida (ej. sf-36)
- Por deseo del paciente
- Si hay pocas posibilidades de seguimiento
Los efectos adversos más frecuentes durante la administración del fármaco son rash cutáneo, fiebre, malestar, tos y taquicardia de leve a moderada y especialmente al principio del tratamiento. Se reducen con infusión más lenta del medicamento o con pre medicación.
En caso de reacción adversa moderada, grave o recurrente por hipersensibilidad relacionada con la administración del fármaco es recomendable el estudio de IgE, de la activación del complemento y/o de la triptasa sérica. (Farmacovigilancia). Está contemplada también la posibilidad de realizar una prueba cutánea con Myozyme® en determinados pacientes con reacción asociada a la perfusión e indicación clara de tratamiento. Por el riesgo que supone es recomendable seguir estrictamente las normas indicadas en el Dossier con informacion de seguridad facilitado por el laboratorio del fármaco y la supervisión de un alergólogo [16].
- Soporte ventilatorio, en aquellos pacientes que lo necesiten. Es preciso tener en cuenta que las alteraciones se producen por debilidad de la musculatura respiratoria y no por broncoespasmo o por hipoxia, con desaturaciones y probable retención de carbónico. Si este hecho se produce habría que valorar la utilización de bipap o respirador volumétrico con mascarilla. En casos más graves puede ser precisa la ventilación invasiva mediante traqueotomía.
- Soporte cardiológico en caso necesario, con inotrópicos, diuréticos, inhibidores de la enzima conversora de angiotensina o betabloqueantes según necesidades y estado de la cardiopatía.
- Soporte nutricional: 25-30% de proteínas, 30-35% de hidratos de carbono, 35-40% de grasas. Puede ser útil suplementar con Alanina (0,14 gr/Kg/dia) [17].
- Fisioterapia: motora, respiratoria.
- Logopedia y foniatría
Tratamiento curativo
Actualmente se están trabajando sobre varias hipótesis para encontrar una solución definitiva para la enfermedad de Pompe. Otras en cambio se han desestimado por su pobre resultado.
- Transplante de médula ósea, utilizado en otras enfermedades de depósito lisosomial no ha sido eficaz en la enfermedad de Pompe. Se desconoce si asociado a otras terapias pudiera ser útil en el futuro.
- En fase de investigación se encuentran: Terapia génica con introducción de vectores virales usando virus adeno-asociados que permitan la infección de la célula y la producción de la enzima por parte de ésta administrada endovenosa o directamente en el músculo [18].
- Chaperonas moleculares [19 y 20].
- Regeneración de tejidos con células madre extraídas de médula ósea [21].
- Inhibidores del sustrato, ensayados con éxito en otras enfermedades de acúmulo.
8.- Seguimiento Clínico
Forma infantil precoz
El seguimiento clínico recomendado para estos pacientes es el siguiente:
- Seguimiento del desarrollo psicomotor usando escalas de desarrollo (Denver o Haizea Llevant).
- Control ecocardiográfico cada 6 meses.
- Control bioquímico: Hemograma, enzimas hepáticas, CPK cada 3 – 6 meses.
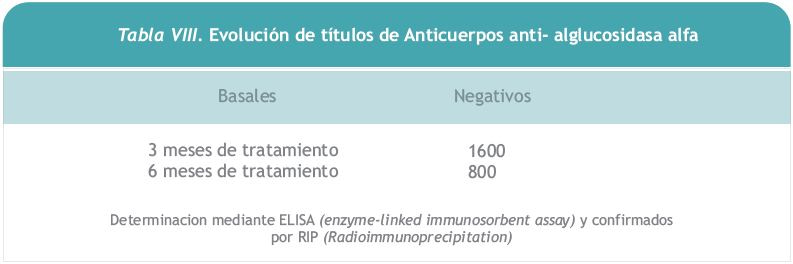
- En caso de terapia enzimática sustitutiva: control de cifras de anticuerpos basales (que deben ser negativos) antes de comenzar la terapia y posteriormente cada 3 meses los 2 primeros años del tratamiento y posteriormente anual).
- Monitorización del tratamiento sustitutivo midiendo el tetra sacárido de glucosa (Glc4) en orina.
Formas tardías
- Escalas de calidad de vida SF-36 o posibilidad de actividades cotidianas como: caminar dentro y fuera del hogar, acostarse o levantarse de la silla o de la cama, ponerse de pie o autocuidado.
- Control bioquímico.
- Seguimiento respiratorio: espirometría, polisomnografía anual.
- Densitometría ósea.
- Audiometría.
- RNM espectroscópica muscular, en caso necesario [22].
9.- Precauciones
Anestesia
La anestesia general puede resultar complicada en los pacientes con enfermedad de Pompe por la presencia de miocardiopatía hipertrófica y su efecto sobre la precarga y postcarga y por la disfunción ventricular que puedan presentar. Los enfermos de Pompe tienen mayor riesgo de isquemia sub-endocárdica y bajo gasto en procedimientos anestésicos. Hay un mayor riesgo de sufrir parada cardio-respiratoria si se utiliza Halotano, Sevofluorano o Propofol. Se recomienda evitar el Suxamethonium por el riesgo de rabdomiólisis con hiperpotasemia. Se considera más seguro el uso de Ketamina y Etomidato al proporcionar estabilidad hemodinámica y se consideran apropiados para la inducción anestésica en pacientes con la enfermedad de Pompe.
Se recomienda, antes de cualquier intervención, ponerse en contacto con el médico anestesista, realizar una ecografía preoperatoria para determinar el grado de la afectación cardíaca, monitorización ECG intra-operatoria, vigilancia y monitorización de potasemia, creatinkinasa y mioglobinuria.
El uso de técnicas de anestesia regional para evitar la intubación con una sedación mínima puede ser buena práctica [23,24 y 25].
Vacunación
En estos pacientes es crucial la prevención de enfermedades respiratorias, ya que puede agravar de forma importante el estado clínico. Deben considerarse población de riesgo, recomendándose la vacunación anual antigripal y tener especial cuidado evitando contacto con personas enfermas por la posibilidad de complicaciones respiratorias severas. Están indicadas también otras inmunizaciones como el Palivizumab para prevenir la infección por virus VRS y la vacuna antineumocócica [26].
Problemas respiratorios durante el sueño
La evaluación respiratoria es obligada. Cuando la edad del paciente lo permita deben hacerse espirometrías tanto en decúbito como en supino, ya que la caída del 25% de la capacidad vital (CV) se considera índice de debilidad diafragmática. En pacientes mayores de 6 años es recomendable el estudio polisomnográfico por la posibilidad de apneas de sueño e hipo ventilación nocturna, especialmente si presentan somnolencia diurna o cefaleas matutinas.
Debe vigilarse también la correcta nutrición para optimizar la función muscular.
10.- Caso Clínico
Varón de 2 meses. Producto de primera gestación, gemelar. Motivo de consulta: rechazo de la alimentación y escasa ganancia ponderal. Hermana asintomática. A la exploración destaca escasa movilidad voluntaria, debilidad, falta de control cervical e hipotonía global. Presentaba sonrisa afectiva, balbuceo,contacto social, fijación y persecución ocular. En resumen retraso motor puro, estando conservadas otras áreas del desarrollo psicomotor.
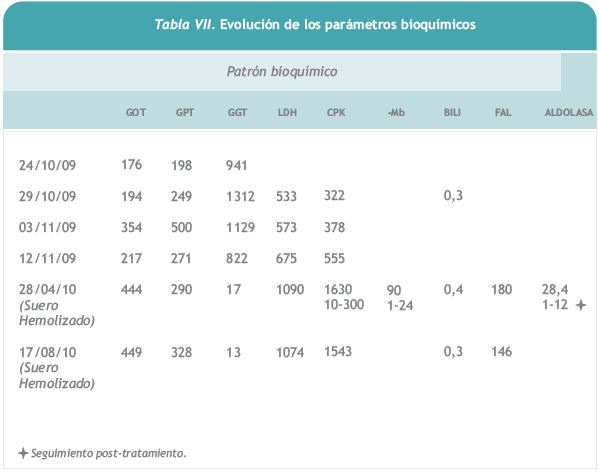
En los exámenes complementarios destaca aumento de GOT, GPT, aldolasa y CPK con bilirrubina normal.
El hemograma y la coagulación fueron normales.
El estudio Electromiográfico mostraba patrón miopático con descargas pseudomiotónicas.
La ecocardiografía reveló la existencia de aumento del tamaño del tabique interventricular sin obstrucción en la salida. La coincidencia de enfermedad muscular periférica, con afectación muy precoz a nivel hepático y cardíaco y que esta afectación fuera
una miocardiopatía hipertrófica orientó el caso a posible enfermedad de Pompe.
Se solicitó estudio en gota seca, usado actualmente como screening por su rapidez.
El resultado fue el siguiente:
- Dosaje enzimas en gota seca:
Alfa-glucosidasa reacción neutra/ácida lisosomal 83 umol/L/h (patológico > o = a 30).
Porcentaje de inhibición de alfa glucosidasa ácida: 90.6 (patológico > o = a 89.0)
Se solicitó estudio genético donde se confirmó la situación de heterocigosidad para el gen GAA de 2 mutaciones tipo missense. (Mutación que altera el significado del codón, codifica para otro aminoácido / mis = mistake = error/).C.1064 T>C (p.L355P) (potencialmente menos severa). C.1209 C>G (p.N403K) –No descrita- (desconocida)
Según consta en el registro internacional, la primera mutación se considera como potencialmente menos severa. La segunda mutación no está descrita hasta el momento por lo que se desconoce cómo se comporta clínicamente.
Inicio tratamiento con alglucosidasa, a 20 mg/Kg intravenoso, sin efectos secundarios relevantes, con una periodicidad quincenal.
Respuesta clínica
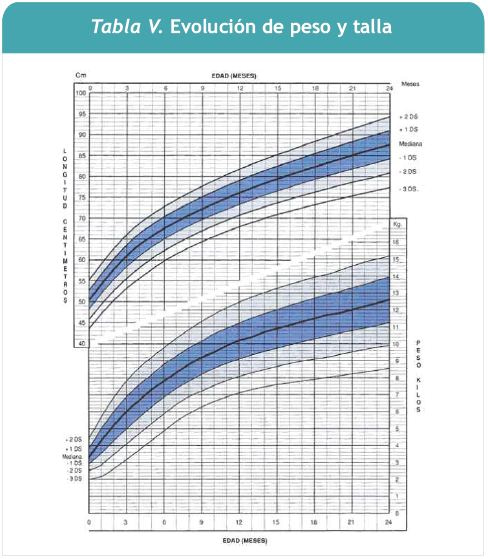
1. Respuesta Somática
El peso y la talla mantienen una curva ascendente, en percentil 45 para peso y algo menor para la talla.
2. Respuesta Neurológica
Clínicamente ha evolucionado favorablemente, con reducción de la hipotonía y consiguiendo progresar en su desarrollo psicomotor.
Al año de vida presentaba solo leve hipotonía y había adquirido la marcha con dos puntos de apoyo. Presenta un leve retraso de lenguaje expresivo con buena comprensión. El área personal social nunca estuvo afectada.
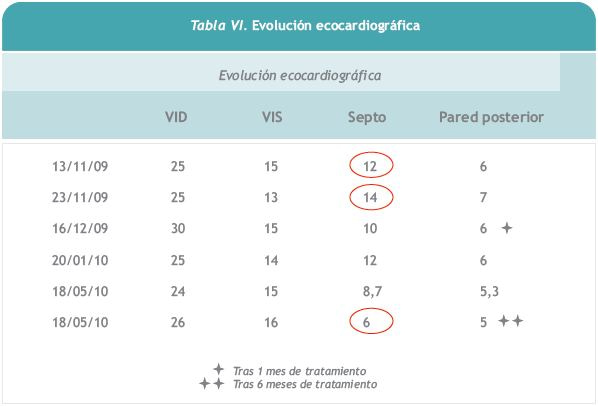
3. Respuesta Cardiológica
Si bien el paciente no llegó a tener clínica cardiológica, los patrones ecocardiográficos eran patológicos, empeorando de forma notable en un corto espacio de tiempo (2 mm de incremento del septo interventricular en solo 10 días). Tras el tratamiento estos valores mejoraron, de forma que a los 6 meses de iniciada la infusión con alglucosidasa ácida, el tamaño del septo interventricular llegó a estar dentro de valores normales para su edad.
4. Respuesta bioquímica
Las determinaciones previas al tratamiento revelaban un daño progresivo hepático y muscular. En los controles posteriores al inicio
del tratamiento se ha observado un incremento de las transaminasas y de la CPK pero no de la GGT, lo que se ha atribuído a una interferencia de los valores al estar el suero hemolizado y con agregados plaquetarios. Si ha bajado de forma considerable la GGT, lo que apoya la posible interferencia y no la progresión de daño hepático.
En cuanto a lo referido en la bibliografía la mejoría en los valores de las transaminasas después de iniciado el tratamiento puede tardar algunos meses [27]. Aún así es recomendable continuar los controles para comprobar si se frena el deterioro hepático y/o se está produciendo un daño tóxico por la propia medicación. En caso de sospecha de relación causa-efecto por la medicación es precisa la notificación como efecto adverso al departamento de farmacovigilancia de Genzyme siguiendo las recomendaciones de la ficha técnica del producto [28].
5. Respuesta inmunológica
Tras las primeras infusiones hubo una respuesta ascendente en la producción de Ig G, con un valor a los 3 meses de 1600. Al ser buena la respuesta clínica y ante la falta de efectos secundarios se siguió administrando la enzima. A los 6 meses de iniciado el tratamiento se solicitó una segunda determinación de anticuerpos bajando estos a la mitad. Hasta el momento no ha presentado efectos adversos por hipersensibilidad.
11.- Discusión
La posibilidad de usar la enzima sustitutiva supone retrasar por algún tiempo la devastadora progresión de la enfermedad de inicio temprano. También es posible mejorar la calidad de vida de estos pacientes. No obstante no supone un tratamiento definitivo, lo cual hace necesario estar atentos a los nuevos tratamientos que actualmente están en fase de investigación. Por otro lado, al ser únicamente de administración intravenosa supone una dependencia de los servicios sanitarios especializados y además la posibilidad de efectos adversos o complicaciones asociadas añadidas a los de la propia enfermedad.
La rápida progresión de la enfermedad hace muy necesaria la precocidad en el diagnóstico, ya que el éxito del mismo depende en gran medida de que éste se aplique antes del deterioro del paciente. Completar el estudio genético es también primordial y no solo ayuda a elaborar el pronóstico por la relación genotipo-fenotipo sino a la elección del tratamiento definitivo cuando este sea posible. El estudio debe ampliarse a los padres y ofrecerles consejo genético.
Referencias Bibliográficas
1. Online mendelian inheritence in man (OMIM). The John Hopkins University.Baltimores.www.nbci.nlm.nih.gov.
2. Pompe, Enfermedad de. SIERE. Sistema de informacion de enfermedades raras en Espa-nol(http://ier.isciii.es/er/prg/er_bus2.asp?cod_enf=2210).Van der Ploeg AT, Reuser AJ. Lysosomal Storage Disease 2. Pompeom disease. Lancet 2008; 372: 1342-53.
3. Reuser AJ et al. Clinical diversity in glycogenosis typeII. Biosynthesis and in situ localization of acid alphaglucosidase in mutant fobroblast. J.Clin Invest 1987;79:1689-99.
4. Hermans MM et al. Twenty-two novel mttations in the lysosomal alpha-glucosidase gene (GAA) underscore the genotype-phenotype correlation in glycogen storage disease type II. Hum Mutat 2004; 23: 47-56.
5. Kroos M, et al. Update of the Pompe. Update of the Pompe disease mutation database with 107 sequence variants and a format for severity rating. Hum Mut 2008; 29: 6, E13- E26.
6. Slonim A et al. Bening course of glycogen storage type II in two brothers:nature or nurture? Muscle Nerve 2006; 33: 571-74.
7. Kingston CP, Sabio Paz V, Solana C. Miocardiopatia hipertrofica neonatal: Una forma de presentación clínica de la enfermedad de Pompe. Arch argent pediatr 2006;104(5): 431-44.
8. Slonin AE et al. Identification of two subtypes of infantile acid maltase deficiency. J. Pediatr 2000; 137: 283-35.
9. Pascual S.I. Enfermedad de Pompe. Diagnóstico y tratamiento de las enfermedades metabólicas 3a Edición. Cap 65. pg 909-19.
10. Jastrzebski M. Short PR intervalo in Pompe di-sease. Journal of Internal Medicine 2009; 266 (6): 571-72.
11. Ansong AK et al. Electrocardiographic response to enzyme replacement therapy for Pompe disease. Genet Med 2006; 8(5): 297-301.
12. Young SP et al. Diagnostic value of urinary and olasma glucose tetrasaccharides in infantile and la-te onset glycogen storage disease type II. Mol Genet Metab 2005; 84:241-42.
13. An Y et al. Glucose tetrasaccharide as a biomar-ker for monitoring the therapeutic response to enzy-me replacement theraphy for Pompe disease. Mol Genet Metab 2005;85: 247-54.
14. Mendelsohn NJ et al. Elimination of antibodies to recombinant enzyme in Pompeom disease. New England Journal of Medicine 2009; 360(2): 194-95.
15. Sun Bet al. Enhanced response to enzyme replacement therapy in Pompe disease: after the induc-tion of immune tolerance. American Journal of Human Genetics 2007;81(5): 1042-49.
16. Berstein Il, Storms WW. Practise parameters for allergy diagnostic testing. Ann Allergy Asthma Im-munol 1995; 75 (6Pt2): 543-625.
17. Bodamer OAF, Halliday D, Leonard JV. The effec-ts of L-alanine supplementation in late-onset glyco-gen storage disease type II. Neurology 2000; 55: 710-12.
18. Sun B et al. Correction of glycogen storage disease type II by an adeno-associated virus vector containing a muscle-specific promoter.Molecular Therapy 2005; 11(6):889-98.
19. Gort L, Coll MJ, Chabas A. Glycogen storage disease type II in Spanish patients: Haigh frecuency of c.1076-1G>C mutation. Molecular Genetics and Metabolism 2007;92 (1-2): 183-87.
20. Porto C et al. The pharmacological chaperone Nbutyldeoxinojirimycin enhanced enzyme replacement therapy in Pompe disease fibroblasts. Molecular Therapy 2009;17(6): 964-71.
21. Douillard-Guilloux G at al. Partial phenotypic correction and immune tolerance induction to enzyme replacement theraphy after hematopoietic stem cell gene transfer of alpha-glucosidase in Pompe disease. Journal of gene Medicine 2009; 11(4): 279-87.
22. Wary C et al. Evaluation of muscle glycogen content by 13C NMR spectroscopy in adult-onset acid maltase deficiency. Neuromuscular disorders 2003; 13 (7-8): 545-53.
23. Ing R et al. Anaesthetic magement of infants with glycogen storage disease type II: a physiological approach. Paediatric Anaesth 2004; 14(6): 514-19.
24. McFarhme HJ, Soni N. Pompeom disease and anaesthesia. Anaesthesia 1986;41(12):1219-24.
25. Rosen K, Broadman L. Anaestesia for diagnostic muscle biopsy in an infant with Pompet disease. Can Anaesth Soc J 1986; 33(6): 790-94.
26. Bembi E et al. Management and treatment of glycogenosis type II. Neurology 2008;71 (Suppl 2): S12-36.
27. Winkel L et al. Enzyme Replacement Therapy in Late-Onset Pompe’s Disease: A Three-Year Follow-up. Ann Neurol 2004; 55: 495–502.
28. Pautas para la administracion en perfusion, notificacion de los acontecimientos adversos y pruebas de inmunogenicidad. SP-V1-23 Mayo 2007.

Más notas de la edición 13
Lee desde Issuu nuestra última edición publicada en Julio 2024, Edición número 155

Notas relacionadas a Variedades de debut temprano en la Enfermedad de...

