
Síndrome hematofagocítico. Reporte de cuatro casos y revisión de la literatura
Pablo Young1, José Peroni1, Bárbara C. Finn1, Julio E. Venditti2, Verónica Preiti3, Eduardo Bullorsky3, Julio E.Bruetman1
1 Servicio de Clínica Médica; 2 Servicio de Patología; 3 Servicio de Hematología.
Hospital Británico de Buenos Aires, Argentina. Rev Med Chile 2011; 139: 224-229
Correspondencia a: Pablo Young Hospital Británico. Perdriel 74 (1280) Buenos Aires, Argentina. Tel: 5411 43096400. Fax: 5411 43043393. E-mail:pabloyoung2003@yahoo.com.ar
La hemofagocitosis puede ser encontrada como un fenómeno aislado en múltiples situaciones como en la anemia hemolítica, enfermedades metabólicas, sepsis y malignidad. El síndrome hemofagocítico reactivo o secundario (SH) o linfohistiocitosis hemofagocítica (LHH) es una entidad clínico-patológica caracterizada por la proliferación sistémica de macrófagos benignos con prominente actividad hemofagocitaria. El SH fue descrito por Chandra y col(1) en 1975 en la población adulta, pero fueron Risdall y col(2) en 1979 quienes lo caracterizaron adecuadamente distinguiéndolo de la variante histiocitosis maligna infantil. Se describen cuatro casos internados en el Hospital Británico entre los años 2004 y 2009, y una revisión de la literatura.
Caso 1
Mujer de 61 años, con antecedentes de hipertensión arterial, hipercolesterolemia, diabetes mellitus tipo 2 no insulino requiriente. Comenzó en diciembre de 2008 con cuadro de mialgias en cinturas escapular y pelviana a predominio proximal más febrícula en contexto de rosuvastatina 20 mg día, asociado a cuadro de odinofagia y cefalea ocasional, a predominio temporal izquierdo, habiendo realizado tratamiento antibiótico con amoxicilina durante una semana con hisopado negativo. Se internó en enero del 2009 dada la persistencia de la sintomatología sumada a fiebre con pirogenemia y dolor pleurítico izquierdo. Su examen físico era normal, sin adenopatías, sin visceromegalias, arterias temporales normales. A nivel del laboratorio, el recuento de glóbulos blancos era de 18.800 por mm3 a predominio polimorfonuclear, el hematocrito 38%, recuento plaquetario 418.000 mm3, creatininemia 0,9 mg/dL, calcemia 8,8 mg/dL, fosfatemia 2,2 mg/dL, uricemia 3 mg/dL, uremia 35 mg/dL, eritrosedimentación (VSG) 102 mm/1 hora, proteína C reactiva (PCR) 16 (VN: menor a 0,3 mg/dL), pruebas hepáticas y electrolitos plasmáticos normales. Electroforesis de proteínas con leve aumento de alfa 2. Sedimento de orina normal. El ECG y la radiografía de tórax fueron normales. Se realizaron tres hemocultivos y un urocultivo que fueron negativos. Se realizó una tomografía axial computada (TAC) de cuello, tórax y abdomen que mostró engrosamiento pleural izquierdo con leve derrame pleural, sin tromboembolismo pulmonar y resto sin particularidades. Inició tratamiento empírico con ceftriaxona cubriendo probable foco respiratorio. El ecocardiograma trans-esofágico no mostró vegetaciones. A pesar del tratamiento antibiótico, continuó con registros febriles aislados, por lo cual aproximadamente a los 20 días inició meprednisona 60 mg/día y gammaglobulina intravenosa en dosis de 400 mg/kg/día por 5 días previo estudios serológicos que se detallan a continuación. Serologías para HIV, sífilis, toxoplasmosis, citomegalovirus (CMV) (serología IgM y antigenemia pp65), Chagas, parvovirus B19, Huddleson, Widal, virus de Epstein Barr (VEB), herpes 6, hepatitis A, B, y C, negativos.
Estudios inmunológicos: Factor antinuclear, Latex, anti DNA, anti Ro y La, RNP, Sm, ANCA c y p, anticuerpos anticardiolipina, CH50, C3 y C4 normales o negativos. Otros estudios: Vitamina B12 y fólico normales. A pesar del tratamiento esteroideo continuó con registros febriles a predominio nocturnos y pirogenemia a lo que agregó en la analítica pancitopenia con plaquetopenia 33.000 mm3, anemia con hematocrito 26%, y leucopenia de 2.000 por mm3. Se solicitó ferritina que fue de 2.000 ug/L (VN: hombres 30-400 y mujeres 15-125), triglicéridos 301 mg/dL y fibrinógeno 109% (normal de 200 a 400%). Se decidió realizar punción aspiración de medula ósea (PAMO) la cual informó: celularidad medular del 50%, hiperplasia megacariocítica con cambios dismórficos, se evidencian fenómenos hemofagocíticos (Figura 1).
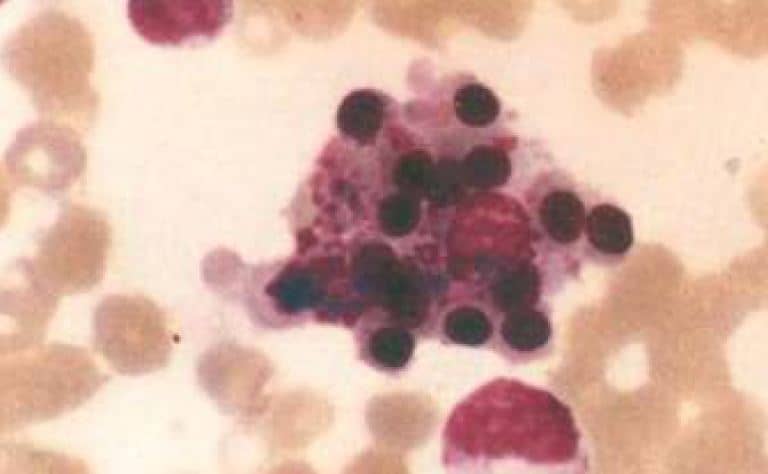
Figura 1. Aspirado de médula ósea con tinción de May Grunwald Giemsa, vista en 100X. Celularidad y progenies conservadas con fenómeno de hemofagocitosis, se observa un macrófago con partículas fagocitadas en su citoplasma y restos nucleares.
En base a dichos hallazgos se interpretó el cuadro como un SH secundario a posible causa infecciosa versus autoinmune. Se decidió iniciar tratamiento con ciclosporina 0,2 cc (200 mg) c/12 h y rotar la metil-prednisolona a dexametasona10 mg c/12 h. La paciente presentó buena respuesta al tratamiento, no constatándose más registros febriles y normalizando el hemograma y las plaquetas. A la semana del egreso se realizó PCR para CMV, siendo esta positiva (> 5000 copias; VN: menor de 500 copias) agregándose al tratamiento previo, ganciclovir, con la consiguiente disminución de carga viral y mejoría del cuadro. Se realizó una disminución gradual de esteroides y se suspendió la ciclosporina a los dos meses de iniciada. Se interpretó el cuadro como un SH secundario a CMV. Luego de 1 año la paciente se encuentra asintomática.
Caso 2
Hombre de 61 años de edad con antecedente de hipertensión arterial que consultó por astenia, adinamia y fiebre de 38,5° axilar de dos semanas de evolución, asociado a la aparición de placas eritematosas con áreas de descamación, pruriginosas en arcada inguinal, axilas y en región periumbilical. Ingresa con signos vitales estables, febril, lesiones cutáneas descriptas, hepatomegalia palpable de tres traveses de dedo y esplenomegalia.
Sin adenopatías. Se realizó biopsia de piel que mostró prurigo urticariano inespecífico. A nivel del laboratorio el recuento de glóbulos blancos era de 3.000 por mm3 a predominio polimorfonuclear, el hematocrito 29%, recuento plaquetario 95.000 mm3, LDH 9.500 U/L, ferritina 1.500 ug/L, VSG 120 mm/1 hora, PCR 5,5 mg/dL, fibrinógeno 108%, triglicéridos 450 mg/dL, hepatograma alterado con TGP 376 UI/mL, TGO 125 UI/mL y FAL 210 mg/dL (VN: TGP: menor a 30; TGO: menor a 32; FAL: menor a 126). Electroforesis de proteínas con hipoalbuminemia y leve aumento de alfa 2. Sedimento de orina normal. Todas las serologías virales e inmunológicas fueros negativas. Se realizaron cuatro hemocultivos y un urocultivo que fueron negativos. Se realizó una tomografía axial computada de cuello, tórax y abdomen que mostró hepatoesplenomegalia y resto sin particularidades.
Se realizó PAMO que mostró hemofagocitosis. Por cumplir seis de los criterios se hace diagnóstico de SH y comenzó terapia con corticoides a altas dosis y gammaglobulina intravenosa en dosis de 400 mg/kg/día por 5 días con desaparición de la fiebre.
A los días de la administración de la gammaglobulina reapareció la fiebre con incremento de esplenomegalia, por lo que se realizó esplenectomía diagnóstica ante la sospecha de proceso linfoproliferativo maligno. La biopsia mostró esplenomegalia congestiva, expansión de pulpa roja, hematopoyesis extramedular, ausencia de neoplasia, signos de hemofagocitosis y la biopsia en cuña del hígado esteatosis leve, colestasis mínima y ectasia sinusoidal. Se comenzó terapia con 2 eEtopósido 150 mg/m . El paciente evolucionó con tricitopenia severa con alto requerimiento transfusional y neutropenia febril prolongada. Nueva PAMO reveló aplasia medular. Desarrolló fallo multiorgánico, coagulopatía por consumo, sangrados masivos y falleció a los 47 días del ingreso.
Caso 3
Hombre de 23 años que vive en Lobos provincia de Buenos Aires que ingresó por fiebre de tres días de evolución y pancitopenia con plaquetopenia 53.000mm3, anemia con hematocrito 29%, y leucopenia de 3 2.500 por mm .Se realizaron hemocultivos y un urocultivo que fueron negativos.
Al examen físico presentó importante inyección conjuntival y ligera esplenomegalia. Los anticuerpos IgM e IgG para Hanta virus fueron positivos y negativas los de leptospira y fiebre hemorrágica argentina, entre otras. Al tercer día de internación desarrolla insuficiencia respiratoria e infiltrados en vidrio esmerilado en la TAC de tórax con lo cual se hace diagnóstico de síndrome pulmonar por Hanta virus. Se le realizó una PAMO que mostró un aumento de macrófagos en 8% con hemofagocitosis.
Se solicitó ferritina que fue de 7.000 ug/L, trigliceridos 401 mg/dL y fibrinógeno 100%. Con todos estos datos se realizó el diagnóstico de SH asociado a Hanta virus. A los 10 días de internación sin tratamiento específico normalizó las citopenias y fue dado de alta.
Caso 4
Hombre de 72 años sin antecedentes de importancia que ingresó por insuficiencia respiratoria y fiebre con diagnóstico de neumonía. Los cultivos y las serologías para virus herpes y CMV fueron negativos, además de todas las serologías al igual que los pacientes previos. Recibió tratamiento antibiótico empírico. A las dos semanas de su ingreso presentó fiebre y empeoramiento de las imágenes radiológicas basales derechas. Se realizó BAL que no mostró crecimiento bacteriano. El paciente desarrolló paulatinamente citopenias de las tres series: Hb: 7 g/L; recuento de leucocitos: 1.600/mm3; plaquetas: 49.000 /mm3. La ferritina fue de 4.000 ug/L, triglicéridos 607 mg/dL, fibrinógeno 111%. Se le realizó una PAMO que mostró un aumento de macrófagos en 10% con hemofagocitosis.
Al examen físico solo presentó ligera hepatoesplenomegalia.
Se le administró metilprednisona a una dosis de 1 g/día por tres días y gammaglobulina en dosis de 400 mg/kg/día por 5 días. Evolucionó con fiebre persistente y requerimiento de asistencia respiratoria mecánica. Las citopenias no mejoraron. Se repitieron cultivos que siempre fueron negativos, a excepción de un BAL que evidenció el crecimiento significativo de una Acinetobacter baumannii multirresistente que recibió tratamiento. Falleció a los 44 días del ingreso con falla multiorgánica.
Discusión
Las histiocitosis han sido clasificadas por el grupo de trabajo de la Sociedad Histiocitaria (www.histio.org/society) en 3 grupos: 1) desórdenes relacionados a células dendríticas; 2) desórdenes relacionados a macrófagos y 3) desórdenes malignos (3). El SH pertenece a la segunda categoría, conjuntamente con el síndrome de activación macrofágica (SAM) y la LHH familiar. El término SAM acuñado por Hadchouel y col4 en 1985; ha sido usado casi exclusivamente para describir el SH en asociación con enfermedades reumáticas, especialmente la artritis reumatoidea juvenil (5).
El SH puede ser primario o secundario, las formas primarias o genéticas se dividen en dos subgrupos, uno es la familiar o enfermedad de Farquhar y el otro son los síndromes asociados a deficiencias inmunes como el síndrome de Chediak Higashi (Tabla 1). En cuanto a la forma familiar, el 80% de los casos ocurre antes del año de edad. Las formas adquiridas o secundarias son principalmente virales y de ellas el VEB y CMV son los agentes más frecuentes, de las causas malignas el linfoma es la más común (Tabla1)5,6.

El cuadro clínico se caracteriza por fiebre prolongada, hepato-esplenomegalia y citopenias. Las linfadenopatías, el eritema cutáneo, la ictericia y los síntomas neurológicos son menos frecuentes.

Típicamente a nivel del laboratorio existe hipertrigliceridemia, niveles de ferritina, transaminasas, bilirrubina, LDH elevados y fibrinógeno bajo (6).
Los cuatro pacientes presentados en esta comunicación fueron estudiados, pero no tratados en forma uniforme. El diagnóstico fue sospechado luego de descartar otras causas más frecuentes y conocidas. Todos ellos presentaron depleción drástica de las tres series sanguíneas. La fiebre fue el signo presente en todos los pacientes y todos ellos presentaron hemofagocitosis en la primera punción de médula ósea, hallazgo que no es universal, al inicio de la enfermedad. Las infecciones virales fueron los desencadenantes del síndrome en 2 pacientes (CMV y Hanta), y en los otros dos casos no se comprobó la causa (7).
Los criterios diagnósticos utilizados son los establecidos en el protocolo HLH- 2004, el que exige 5 de 8 criterios clínicos, de laboratorio e histopatológicos (Tabla 2)8. La fiebre y la esplenomegalia son los criterios clínicos. A las citopenias, se agregan la hipertrigliceridemia > 265 mg/dL e hipofibrinogenemia 2% de todas las células nucleadas de la médula ósea sean histiocitos con Hemofagocitosis (9).

Los hallazgos clínicos durante la fase aguda del SH pueden ser explicados como consecuencia de la acción de citoquinas originadas presumiblemente en macrófagos y células T activadas.
La anormalidad inmunológica más frecuente es la alteración global de la función citotóxica. La función de las células NK está marcadamente disminuida o ausente y la actividad citotóxica de los CD8+ también es defectuosa recuperánd ose luego del tratamiento. Muchos marcadores de la activación de los macrófagos (ferritina, b2 microglobulina, enolasa neuronoespecífica) y citoquinas (interferón gamma, factor de necrosis tumoral alfa e interleuquinas) están aumentadas durante el SH. Frecuentemente se detectan altas concentraciones de la cadena alfa del receptor soluble IL-2 (sCD 25), valores superiores a 2.400 u/mL es un nuevo criterio diagnóstico y se asocia con peor pronóstico (6,8,10).

La sepsis grave se asocia con fallo multiorgánico (FMO). La relación entre SH y FMO no es clara (11). Reiner reportó 4 casos de FMO entre 23 casos de SH (12). Recientemente Kleinert y col. reportaron en nuestro país cuatro casos mortales en el contexto de la terapia intensiva (13). Ambas entidades comparten desencadenantes infecciosos entre los que se encuentran virus, bacterias, hongos y parásitos. En ambas está presente un nivel aumentado de citoquinas. Si bien esto no es prueba de su vinculación, sugiere que están asociadas a un estado proinflamatorio.
La respuesta al tratamiento es en general peor en los adultos con SH por razones no conocidas totalmente (6). Los objetivos del tratamiento son suprimir la hiperinflamación responsable de los síntomas y destruir a las células infectadas presentadoras de antígeno. Según el protocolo de la Sociedad Histiocitaria revisado en el año 2004 (HLH-2004), la terapéutica inicial consiste en corticoesteroides (CE), preferentemente dexametasona que cruza la barrera hematoencefálica mejor que la prednisolona, ciclosporina A (CSA) y etopósido (8). Los CE son citotóxicos para los linfocitos e inhiben la expresión de citoquinas, la CSA impide la activación de los linfocitos, el etopósido tiene alta actividad en las enfermedades histiocitarias y es particularmente útil en el SH por VEB. En pacientes con síntomas menos graves, la asociación de CE e inmunoglobulinas puede ser suficiente (14). Dos grandes series han mostrado resultados promisorios con el uso de gammaglobulina endovenosa (GGEV), especialmente en el SH secundario a infecciones (14,15).
Debe enfatizarse que el tratamiento dirigido solamente contra el patógeno no es suficiente, con la posible excepción de la leishmaniasis, en la cual el tratamiento con anfotericina B suele ser curativo.
Tres de nuestros pacientes recibieron GGEV y dos fallecieron sin poder determinar la causa del SH. Recientemente se ha ensayado con éxito el tratamiento con daclizumab (un anticuerpo monoclonal anti-CD25), en un paciente con SH dependiente de esteroides (16).
En conclusión creemos que el médico general tiene que tener un alto índice de sospecha, tener en mente los criterios diagnósticos del SH para instaurar a tiempo un tratamiento que salve la vida.

Referencias Bibliográficas
1. Chandra P, Chaudhery SA, Rosner F, Kagen M. Transient histiocytosis with striking phagocytosis of platelets, leukocytes and erythrocytes. Arch Intern Med 1975; 135:989-91.
2. Risdall RJ, McKenna RW, Nesbit ME, Krivit W, Balfour HH Jr, Simmons RL, et al. Virus associated hemophagocytic syndrome: A benign histyocitic proliferation distinct from malignant histiocytosis. Cancer 1979; 44:993-02.
3. Favara BE, Feller AC, Pauli M, Jaffe ES, Weiss LM, Arico M, et al. Comtemporary classifi cation of histiocytic disorders. The WHO Committee on histiocytic/Reticulum cell proliferations. Reclassifi cation Working Group of the Histiocytic Society. Med Pediatr Oncol 1997;157-66.
4. Hadchouel M, Prieur AM, Griscelli C. Acute hemorrhagic, hepatic and neurologic disease in juvenile rheumatoid arthritis. Possible relationship with drugs or infection. J Pediatr 1985; 106: 561-6.
5. Fukaya S, Yasuda S, Hashimoto T, Oku K, Kataoka H, Horita T, et al. Clinical features of haemophagocytic syndrome in patients with systemic autoimmune diseases:analysis of 30 cases. Rheumatology(Oxford) 2008;47: 1686-91.
6. Janka GE. Haemophagocytic syndromes. Blood Reviews 2007; 21:245-53.
7. Lee JJ, Chung IJ, Shin DH, Cho SH, Cho D, Ryang DW, et al. Hemorrhagic Fever with Renal Syndrome Presenting with Hemophagocytic Lymphohistiocytosis. Emerg Infect Dis 2002; 8: 209-10.
8. Henter JI, Horne A, Arico M, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistio-cytosis. Pediatr Blood Cancer 2007; 48:124-131.
9. Wong KF, Chan JKC. Reactive hemophagocytic syndrome: a clinicopathologic study of 40 patients in an oriental population. Am J Med 1992; 93: 177-80.
10. Egeler RM, Shapiro R, Loechelt B, Filipovich A. Characteristic inmune abnormalities in hemophagocytic lymphohistiocytosis. J Pediatr Hematol Oncol 1996; 18:340-5.
11. Gauvin F, Toledano B, Champagne J, Lacroix J. Reactive hemophagocytic syndrome presenting as a component of multiple organ dysfunction syndrome. Crit Care Med 2000; 28: 3341-45.
12. Reiner AP, Spivak JL. Hematophagic histiocytosis: a report of 23 new patients and a review of the literature. Medicine (Baltimore) 1988; 67: 369-88.
13. Kleinert MM, Garate G, Osatnik J, Cicco J, Hunter B, Soria EJ. [Síndrome hemofagocítico reactivo en pacientes graves. Comunicación de 4 casos]. Medicina (Bs As) 2007; 67: 49-52.
14. Larroche C, Bruneel F, Andre MH, Bader-Meunier B, Baruchel A, Tribout B, et al. Intravenously administered gammaglobulins in reactive hemophagocytic syndrome. Multicenter study to assess their importance by the immunoglobulins group of experts of CEDIT of the APHP. Ann Med Interne (Paris) 2000; 151: 533-9.
15. Emmenegger U, Schaer DJ, Larroche C, Neftel KA. Haemophagocytic syndromes in adults: current concepts and challenges ahead. Swiss Med Wkly 2005; 135: 299-314.
16. Olin RL, Nichols KE, Naghashpour M, et al. Successful use of the anti – CD25 antibody daclizumab in an adult patient with hemophagocytic lymphohistiocytosis. Am J Hematol 2008; 83: 747-49.


Más notas de la edición 21
Lee desde Issuu nuestra última edición publicada en Octubre 2025, Edición número 170

Notas relacionadas a Síndrome hematofagocítico. Reporte de cuatro...

