
Nefropatía C1q: diagnóstico inusual de síndrome nefrótico en la infancia. Reporte de caso
C1q nephropathy: unusual diagnosis of nephrotic syndrome in childhood. Case report
CASO CLÍNICO
Aura Mearlyn Basabe Ochoa1
http://orcid.org/0000-0003-4172-3056
Avelina Victoria Troche Hermosilla1
http://orcid.org/0000-0002-2954-9836
Marlene Martínez Pico1
http://orcid.org/0000-0001-5769-0952
1Instituto de Previsión Social, Hospital Central “Dr. Emilio Cubas”, Servicio de Nefrología, Unidad de Nefrología Pediátrica. Asunción, Paraguay
Revista del Nacional (Itauguá)
Print version ISSN 2072-8174 – Rev. Nac. (Itauguá) vol.12 no.2 Itauguá Dec. 2020 – https://doi.org/10.18004/rdn2020.dic.02.124.129 – Avda. Gral. Marcial Samaniego. Itauguá Guazú. Itauguá, Paraguay. Teléf. 595-294-321-450/4 – hn.biblioteca@gmail.com
Resumen
La nefropatía C1q es una glomerulopatía poco comprendida y subdiagnosticada. Se define por un patrón de inmunfluorescencia dominante o codominante de positividad para C1q, con depósitos electrodensos en mesangio, en ausencia de serología y clínica de lupus eritematoso sistémico. Clínicamente se manifiesta con proteinuria severa o de rango nefrótico, en ocasiones hematuria e hipertensión arterial. Histológicamente presenta morfología variada. Usualmente se manifiesta como síndrome nefrótico corticodependiente o corticoresistente con mala respuesta al tratamiento inmunosupresor y evolución a la cronicidad. Se presenta el caso clínico de un niño diagnosticado con nefropatía C1q, a quien se indicó biopsia renal por cuadro de síndrome nefrótico corticorresistente, con serología negativa y ausencia de datos clínicos para lupus eritematoso sistémico. La intervención oportuna y el manejo temprano permiten enlentecer su evolución a la cronicidad.
Palabras clave: nefropatía C1q, síndrome nefrótico, proteinuria, hematuria
Abstract
C1q nephropathy is a poorly understood and underdiagnosed glomerulopathy. It is defined by a dominant or codominant immunfluorescence pattern of C1q positivity, with electrodense deposits in the mesangium, in the absence of serology and symptoms of systemic lupus erythematosus. Clinically, it manifests with severe proteinuria or nephrotic range, occasionally hematuria and arterial hypertension. Histologically it presents varied morphology. It usually manifests as a corticodependent or cortico-resistant nephrotic syndrome with poor response to immunosuppressive treatment and evolution to chronicity. We present the clinical case of a child diagnosed with C1q nephropathy, who underwent a renal biopsy due to corticosteroid-resistant nephrotic syndrome, with negative serology and absence of clinical data for systemic lupus erythematosus. Timely intervention and early management slow down its progression to chronicity
Key words: C1q nephropathy, nephrotic syndrome, proteinuria, hematuria
Introducción
La nefropatía C1q (NC1q) es una glomerulopatía idiopática que habitualmente se presenta en niños y adultos jóvenes1. Es una entidad poco comprendida y subdiagnosticada, con muy baja prevalencia a nivel mundial que varía de 0.2 a 2.5 %, esta prevalencia aumenta en la población pediátrica, siendo de 2.1 a 6 %, y hasta16 % en niños con síndrome nefrótico2. Fue descripta por primera vez como una entidad clínico-patológica distinta en 1985, por Jenette y Hipp1 y se define histológicamente como un patrón de inmunfluorescencia dominante o codominante de positividad para C1q, con depósitos electrodensos en mesangio, en ausencia de serología y clínica de lupus eritematoso sistémico (LES).
En la infancia, en el síndrome nefrótico primario, el hallazgo más frecuente en las biopsias renales es la enfermedad de cambios mínimos, y en los adultos, la nefropatía membranosa, aunque menos frecuentemente se puede encontrar glomeruloesclerosis focal y segmentaria o glomerulonefritis membrano proliferativa.
Clínicamente la NC1q se manifiesta con proteinuria severa o de rango nefrótico, y en ocasiones con hematuria e hipertensión arterial (HTA). Histológicamente presenta morfología variada, desde enfermedad de cambios mínimos hasta glomeruloesclerosis focal y segmentaria, siendo éste último el hallazgo más frecuente. Algunos autores la consideran como una enfermedad de transición entre los cambios mínimos y la glomerulosclerosis segmentaria y focal (GEFS)1,2. Usualmente se manifiesta como síndrome nefrótico corticodependiente o corticoresistente con mala respuesta al tratamiento inmunosupresor y evolución a la cronicidad. La combinación de glucocorticoides con ciclofosfamida, azatioprina, micofenolato mofetil, tacrolimus y rituximab también han mostrado resultados en algunos estudios6.
En este trabajo se describe el caso de un niño que presentó síndrome nefrótico corticorresistente, a quien se le diagnosticó NC1q mediante la biopsia renal y que presentó buena respuesta a la ciclosporina. Es importante considerar este diagnóstico en todo paciente con síndrome nefrótico resistente a los corticoides, a fin de dirigir el tratamiento según los hallazgos histológicos, de lo cual además dependerá su pronóstico.
Reporte de caso
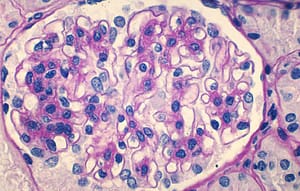
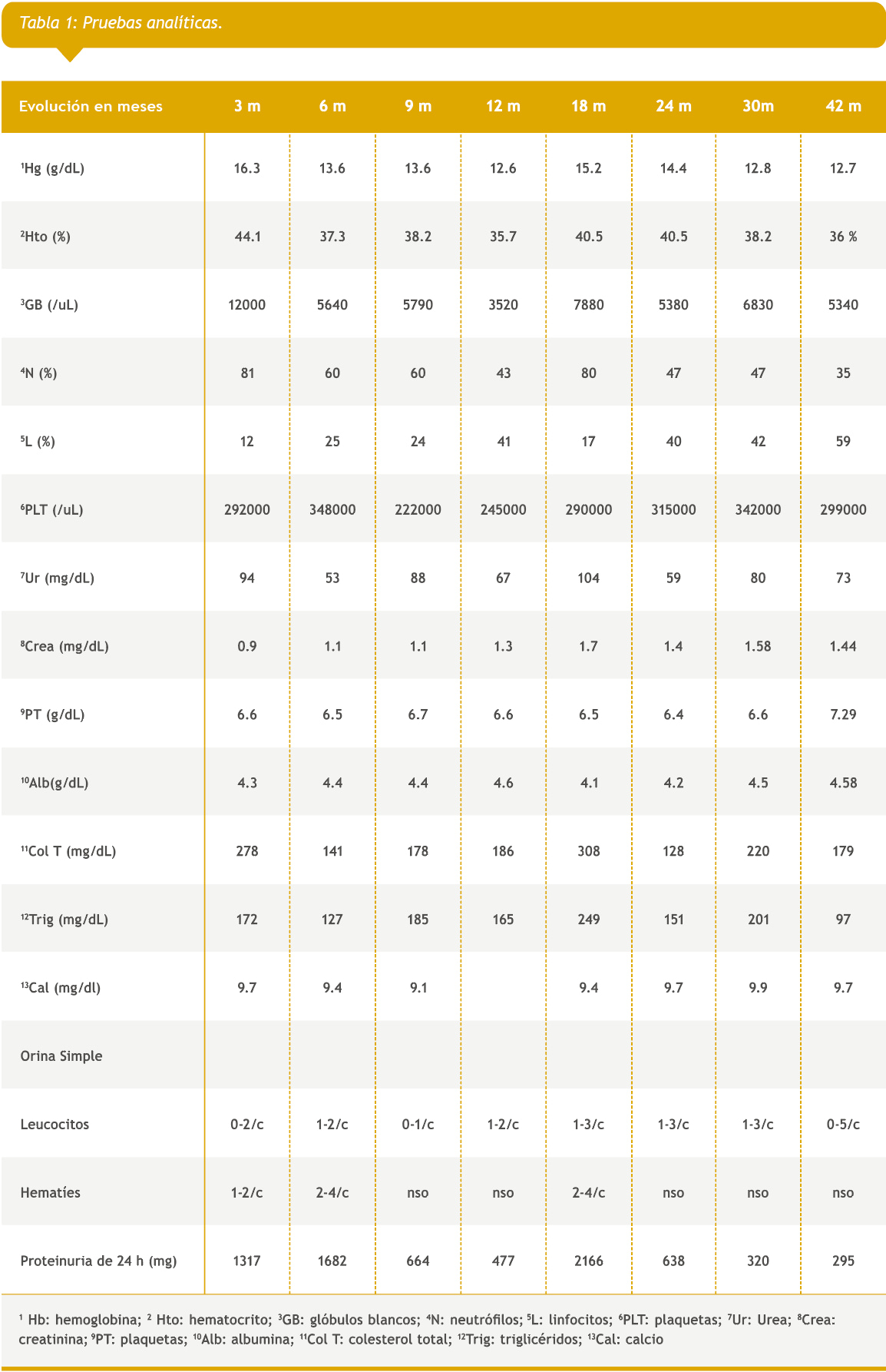
Paciente masculino de raza blanca que al diagnóstico presentaba 8 años. Procedente de San Lorenzo, con antecedentes de cirugía de liberación de sindactilia en ambas manos en 2010. En mayo de 2017 ingresa al servicio de urgencias de nuestro hospital. Presentó edemas, oliguria, dolor abdominal y vómitos. En el examen físico se constató deformidades en manos y pies (antecedente de cirugía de liberación de sindactilia en manos, y sindactilia en ambos pies), edema bipalpebral y en miembros inferiores, que dejó godet (++), cifras de presión arterial por encima del percentil 95 para edad. Eupneico, resto del examen físico sin alteraciones. Posteriormente al correlacionar con la analítica de laboratorio se realiza diagnóstico de síndrome nefrótico, HTA e insuficiencia renal aguda. El análisis de sangre reveló urea 66 mg/dL, creatinina 1,15 mg/dL, colesterol total 298 mg/dL y triglicéridos de 172mg/dL, las proteínas totales 5.9 mg/dL y albúmina 3.9 mg/dL, la hemoglobina glicada fue normal. Las serologías para virus (VIH, virus de hepatitis B y C) fueron normales. El estudio inmunológico anticuerpo anti-nuclear (ANA), anticuerpo anti ADN (ANTI DNA), complementos, antiestreptolisina O (ASTO), dosaje de inmunoglobulina A (IgA) e inmunoglobulina G (IgG), anticuerpo anticitoplasma de los neutrófilos (ANCA) fueron normales. El sedimento urinario mostró proteínas +, leucocitos mayor a 30 por campo y hematíes mayor a 100 por campo. Presentó proteinuria de 24 horas de 2175 mg y su aclaramiento de creatinina fue de 74,37 mL / min/1,73 SC. En la ecografía renal se encontró ambos riñones de tamaño disminuido, con ecogenicidad aumentada, espesor conservado con relación cortico-medular disminuida. Se inició tratamiento con prednisona 60 mg día por 8 semanas persistiendo la proteinuria, por lo que se le administró 3 bolos de metilprednisolona sin respuesta al tratamiento. Se indicó biopsia renal que informó en la microscopía óptica, lesiones proliferativas segmentarias de hipercelularidad endocapilar con aumento de células mesangiales con esclerosis segmentaria con obliteración de luces capilares y fibrosis mesangial en ovillos. En la inmunofluorescencia (IF) presentó imágenes positivas de moderada intensidad de IgG y C1q (++) en mesangio, de intensidad leve (+) en asas capilares focales y segmentarias, todas granulares y de IgM e IgA + en mesangio, con C3 y fibrinógeno negativo. Ante ausencia de serología y clínica de LES, se diagnostica NC1q y se inició tratamiento endovenoso con ciclofosfamida 1 gramo/m2 durante 6 meses, seguido de tratamiento de mantenimiento con micofenolato mofetil (MMF) 1g /día más prednisona por 8 meses, continuando sólo con MMF; la proteinuria (PTU) en ese momento fue de 477 mg/24 h. Al mes, presentó recaída con PTU de 2166 mg/24 h, por lo cual se intentó remisión aumentando dosis de MMF a 2 g/día y reiniciando prednisona a 60 mg/día, sin respuesta a las 8 semanas, por lo que se decidió iniciar ciclosporina 150 mg m2/día en 2 dosis, manteniendo un nivel sanguíneo entre 100 a 200 ng/mL, con lo cual presentó descenso progresivo de PTU. Se suspendió ciclosporina a los 16 meses de haber iniciado el tratamiento, manteniendo valores de proteinuria menores a 400 mg / día. Actualmente está en tratamiento sólo con enalapril 2.5 mg /día, y con valores de urea y creatinina mejorados en comparación con los valores que presentaba antes de iniciar tratamiento con ciclosporina. (Tabla 1)
Discusión
Para el diagnóstico de NC1q se requiere la presencia de C1q en el mesangio con una intensidad >2+ (en una escala de 0+ a 4+) en la microscopía de inmunofluorescencia o inmunohistoquímica, la que puede ser aislada o acompañarse de inmunoglobulinas u otras fracciones del complemento en forma codominante, además de la presencia de depósitos densos mesangiales y/o paramesangiales en conjunto con alteraciones pedicelares significativas(4, 5, 7). En nuestro paciente se llegó al diagnóstico por los hallazgos histológicos, la ausencia de clínica y la serología negativa para LES.
Clínicamente la nefropatía C1q se presenta de forma variada, pudiéndose manifestar como hematuria macroscópica, síndrome nefrítico, síndrome nefrótico, insuficiencia renal aguda, insuficiencia renal crónica, glomerulonefritis rápidamente progresiva, síndrome urémico-hemolítico, vasculitis o hipertensión arterial7,8,9.
El tratamiento depende de la presentación clínica y de los hallazgos morfológicos en la biopsia renal. Algunos estudios sugieren que la nefropatía C1q es clínicamente más agresiva en comparación con otras glomerulopatías, y en mayor proporción en los pacientes corticorresistentes10. Los pacientes nefróticos con NC1q, si bien logran obtener la remisión con corticoides, presentan recaídas frecuentes, se hacen esteroides dependientes o resistentes, debiendo agregarse inmunosupresores como ciclofosfamida, ciclosporina, tacrolimus, micofenolato mofetil o rituximab, para mantener la remisión5,11. Nuestro paciente presentó evolución tórpida, por lo cual requirió varias de estas drogas inmunosupresoras para reducir las dosis de esteroides, respondiendo mejor a la ciclosporina. La limitación de este trabajo es que en nuestro país no contamos con microscopio electrónico, ni se realizan estudios genéticos, los cuáles serían sumamente necesarios para el diagnóstico en este tipo de pacientes.
Conclusión
Nuestro paciente presentó un síndrome nefrótico corticorresistente de evolución tórpida con las características histológicas de NC1q. Esta nefropatía es de difícil diagnóstico por su presentación clínica variable. La intervención oportuna y el manejo temprano permiten enlentecer su evolución a la cronicidad. Quedan aún por descubrir muchos aspectos de esta rara entidad, por lo que el tratamiento es un desafío por la falta de evidencia.
Referencias Bibliográficas
1. Jennette JC, Hipp CG. C1q nephropathy: a distinct pathologic entity usually causing nephrotic syndrome. Am J Kidney Dis 1985; 6: 103-110.
2. Markowitz GS, Schwimmer JA, Stokes MB, Nasr S, Seigle RL, Valeri AM, et al. C1q nephropathy: a variant of focal segmental glomerulosclerosis. Kidney Int 2003;64(4);1232- 40
3. Vizjak A, Ferluga D, Roži? M , Hvala A, Lindi? J, Kersnik Levart T, et al. Pathology, clinical presentations, and outcomes of C1q Nephropathy. J Am Soc Nephrol. 2008;19(11):2237-44.
4. Hisano S, Fukuma Y, Segawa Y, Niimi K, Kaku Y, Hatae K, et al. Clinico pathologic correlation and outcome of C1q nephropathy. Clin J Am Soc Nephrol 2008; 3(6): 1637- 43.
5. Wenderfer SE, Swinford RD, Braun MC. C1q nephropathy in the pediatric population: pathology and pathogenesis. Pediatr Nephrol 2010; 25(8): 1385-96.
6. Devasahayam J, Erode-Singaravelu G, Bhat Z, Oliver T, Chandran A, Zeng X, et al. C1q Nephropathy: The Unique Underrecognized Pathological Entity. Anal Cell Pathol (Amst). 2015;2015:490413. doi: 10.1155/2015/490413
7. Maliakkal JG, Liapis H, White AJ, Ahn S-Y. C1q nephropathy in the setting of granulomatosis with polyangiitistreated with tacrolimus. Clin Kidney J. 2014;7(5): 499-500. doi: 10.1093/ckj/sfu087
8. Kanodia KV, Vanikar AV, Shah PR, Kute VB, Feroz A, Suthar K, et al. C1q nephropathy with acute hemolytic uremic syndrome. Saudi J Kidney Dis Transpl. 2012; 23(3): 556-8.
9. Bhowmik DM, Jain S, Dinda AK, Sharma A, Mahajan S, Agarwal SK. C1q nephropathy presenting as nephritic-nephrotic syndrome. Saudi J Kidney Dis Transpl . 2011;22(3):561-563.
10. Gunasekara VN, Sebire NJ, Tullus K. C1q nephropathy in children: clinical characteristics and outcome. Pediatr Nephrol 2014;29(3):407-13.
11. Muorah M, Sinha MD, Horsfield C, O’Donnell PJ. C1q nephropathy: a true immune complex disease or an immunologic epiphenomenon? NDT Plus 2009; 2(4): 285-91. doi: 10.1093/ndtplus/sfp055.
Autor correspondiente: Aura Mearlyn Basabe Ochoa. Instituto de Previsión Social, Hospital Central “Dr. Emilio Cubas”, Servicio de Nefrología, Unidad de Nefrología Pediátrica. Asunción, Paraguay Correo electrónico:mearlyn@yahoo.com
Basabe Ochoa AM: Conceptualización – Ideas, Conservación de datos, Investigación, Metodología, Administración de proyectos y recursos. Troche Hermosilla AV: Supervisión, validación, Redacción – revisión y edición. Martínez Pico M: Supervisión, validación, Redacción – revisión y edición
Este es un artículo publicado en acceso abierto bajo una licencia Creative Commons


Más notas de la edición 140
Lee desde Issuu nuestra última edición publicada en Octubre 2025, Edición número 170

Notas relacionadas a Nefropatía C1q: diagnóstico inusual de...


