



FIBROSIS QUÍSTICA Test del Sudor – Importancia del método de referencia
Laboratorio Ferreiro a través del Área de Enfermedades Metabólicas asiste en el diagnóstico de Fibrosis Quística (FQ) brindando a Río Cuarto y zona de influencia su profesionalidad, equipamiento y experiencia para la determinación de cloruros mediante el Test o Prueba del Sudor.
Además ofrece y de modo complementario la posibilidad del panel con otros diferentes exámenes para completar el diagnóstico de FQ en sus distintas expresiones.
La Fibrosis Quística (FQ) es una de las enfermedades genético-hereditarias mortales más frecuentes en la raza blanca aunque está descripta en todas las etnias; esta patología multiorgánica posee una incidencia estimada entre uno cada 2500 – 3000 recién nacidos vivos. Se transmite de manera autosómica recesiva, se calcula que una de cada 25 personas es portadora el gen defectuoso de la FQ y que una pareja de portadores tiene un 25% de probabilidad de engendrar un hijo con FQ por embarazo.
Es una enfermedad caracterizada por una disfunción de las glándulas exócrinas, expresando insuficiencia pancreática y bronconeumonía crónica, donde las personas afectadas producen un moco espeso y viscoso provocando obstrucción de los órganos donde se localiza.
Haciendo un poco de historia, en 1934 Andersen (1) describió los hallazgos clínicos y anatomo-patológicos de los enfermos con FQ denominándose por primera vez Fibrosis Quística del páncreas. En 1944, Farber (2) denominó el cuadro como Mucoviscidosis y lo detalló como enfermedad multiorgánica, observando que eran hechos secundarios al bloqueo de los conductos excretores por lo espeso de las secreciones. Di Sant Agnese y colaboradores (3) en 1953 explicaron el aumento de la concentración de Cloruros eliminado por el sudor, constituyendo este descubrimiento la prueba diagnóstica más importante de apoyo a la sospecha clínica de la enfermedad (Prueba del Sudor).
En 1959 Gibson y Cooke (4) desarrollaron la determinación de electrolitos en sudor obtenido por iontoforesis con pilocarpina.
Mucho más cerca, en 1985 se identificó y localizó la mutación responsable de la FQ en el brazo corto del cromosoma y en agosto de 1989 Tsui y Collins (5-6), analizando la secuencia de cADN normal y de pacientes con FQ, comunicaron el descubrimiento del gen causante de la enfermedad ,CFTR, demostrando simultáneamente que la mutación más común es la delección de tres pares de bases que codifican un único aminoácido (fenil alanina), mutación DF508.
Estudios realizados en nuestro país, Argentina, han mostrado un amplio espectro de alteraciones en el gen CFTR. La mutación FD508 está presente en casi el 60% de los cromosomas FQ, de las 52 mutaciones diferentes descriptas, frecuencia similar a las encontradas en el Sur de Europa.
Fisiopatología
A nivel de la membrana apical de las células epiteliales secretoras, la proteína CFTR no funciona adecuadamente como canal de cloro ni como regulador de los canales rectificadores exteriores del cloro y sodio, siendo ésta la primera disfunción que genera la enfermedad, explicando de este modo porque los enfermos de FQ tienen sudor tan salado, porque necesitan enzimas pancreáticas cuando presentan insuficiencia pancreática, porqué se infectan crónicamente y pueden desarrollar cirrosis biliar y diabetes mellitus (24) entre otras complicaciones.
Pesquisa Neonatal
En los últimos años la pesquisa neonatal de FQ sufrió un cambio muy importante en relación con su aceptación, difusión e implementación sistemática dentro de los Programas de Detección Neonatal de Enfermedades Congénitas. Si bien en Argentina existe legislación que determina la obligatoriedad de la Pesquisa Neonatal, aún hoy no hay un esquema definido, con lo que se efectúa en su mayoría a demanda y sin un criterio unificado. El descubrimiento de que la Tripsina se hallaba anormalmente elevada a edades tempranas en pacientes con FQ , llevó a su utilización en el diagnóstico precoz mediante el método de TIR (TRIPSINA INMUNORREACTIVA). Con el posterior hallazgo del gen responsable de la FQ se establecieron distintos protocolos de cribaje sólo con TIR o TIR/ADN. El screening neonatal con TIR tiene buena eficiencia diagnóstica, con una sensibilidad del 85% y una especificidad del 99.6%, pero a pesar de esto el valor predictivo positivo de la TIR sola es relativamente bajo, mientras que las estrategias que combinan TIR con una segunda prueba mejoran significativamente la validez diagnóstica. Revisiones sistemáticas del año 2006 muestran una mejoría significativa en sobrevida media a favor de los grupos diagnosticados por pesquisa neonatal.
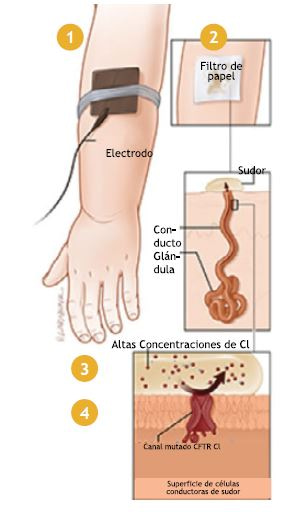
1. El electrodo lleva la pilocarpina hacia la piel.
2. El sudor es recolectado en el filtro de papel.
3. Se realiza un test de Cloruro en el sudor.
4. Altas concentraciones de Cloruro pueden deberse a una mutación CFTR
Diagnóstico Molecular
Es importante destacar que siendo la Fibrosis Quística una enfermedad autosómica recesiva, la detección de dos alelos mutados (una mutación en cada copia del gen CFTR) es diagnóstico de certeza. Sin embargo, la no detección de mutaciones no excluye la patología de FIBROSIS QUÍSTICA porque, como se ha visto, los estudios disponibles se limitan en general al análisis de las mutaciones más frecuentes.
La demostración de 2 mutaciones en personas con fenotipos atípicos y valores de cloruros en sudores intermedios o normales reafirma la necesidad de realizar análisis moleculares. Esta información es crucial para la confirmación diagnóstica y el asesoramiento genético.
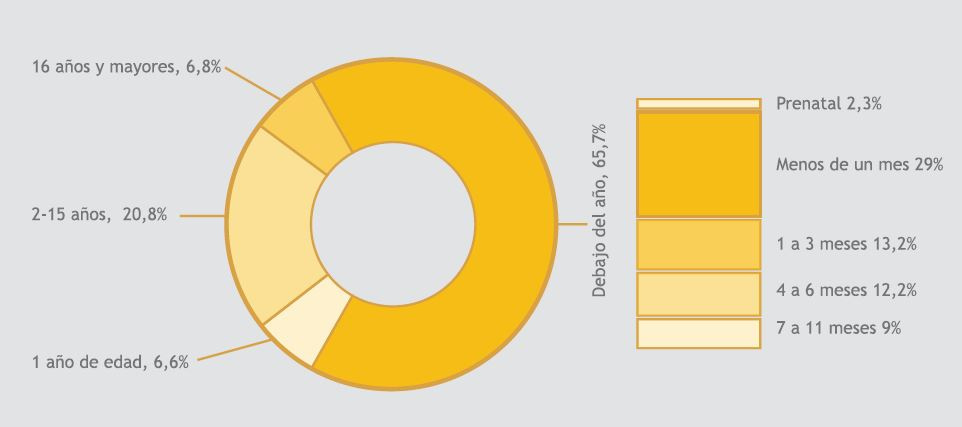
Edades de diagnóstico para todas las personas con F. Q en el Registro 2013 (Segunda imagen a la derecha).
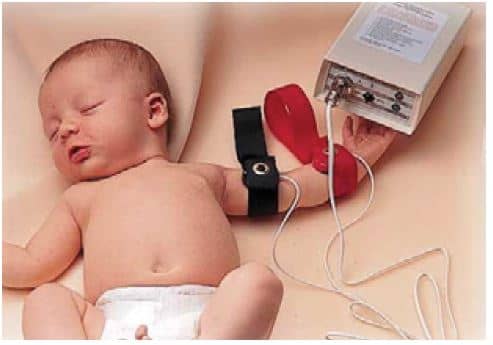
Test Del Sudor
Exámen fundamental para la comprobación del diagnóstico. Se debe realizar después del primer mes de vida y de ser positivo se mantiene como tal en el tiempo. Para su correcta realización el único método aceptado universalmente como fiable es el Método de Gibson Cooke (TEST CUANTITATIVO DE IONTOFORESIS CON PILOCARPINA), considerado como el Gold Standard para el diagnóstico de FIBROSIS QUÍSTICA, cualquier otro test debe ser comparado con éste.
La prueba consiste en la estimulación de las glándulas sudoríparas mediante iontoforesis con pilocarpina, recolección del sudor generado y posterior cuantificación de la concentración de cloro presente en el mismo.
Se obtienen resultados diagnósticos con la obtención de valores mayores de 60 meq/L de Cloruros en dos muestras consecutivas.

Para su correcta realización y resultados confiables es imprescindible destacar que el procedimiento sea llevado a cabo con una metodología estandarizada y por un profesional con la debida capacitación y experiencia.
El test del sudor da cuenta del 98% de los casos de FIBROSIS QUÍSTICA, restando un 2% de formas leves con examen límite o normal.
El GOLD STANDARD metodológico para establecer el diagnóstico actualmente aún sigue siendo el TEST DEL SUDOR.
Gracias a los avances científicos de las últimas décadas,- importantes y novedosos logros en los métodos diagnósticos y en el manejo y tratamiento de los pacientes- se han alcanzado notables mejorías en la calidad y esperanza de vida para estos pacientes, inimaginable tan solo pocas décadas atrás.
REFERENCIAS BIBLIOGRÁFICAS
1-Andersen DH. Cystic fibrosis of the páncreas and its relation to celiac disease:A clinical and pathological study.Am J Dis Child 1938;56:344-349
2-Farber S. Pancreatic function and disease in early life.V.Pathologic changes associated with pancreatic insufficiency in early life.Arch Pathol 1944;37:238-243
3-Di Sant Agnese PA,Darling RC, Perera GA, Shea E.Abnormal electrolyte composition of sweat in cystic fibrosis of the páncreas,its clinical significance and relatioship to the disease.Pediatrics 1953;12:549-563
4-Gibson LE, Cooke RE. A test for concentration of electrolytes in cystic fibrosis utilizing pilocarpine by iontophoresis. Pediatrics 1959;23:545-563
5-Rommens J,Ianuzzi M.,Kerem B,Drumm M,Melmer G et al.Identificationof the cystic fibrosis gene:Chromosome Walking and Jumping.Science 1989;245:1059-1065
6-Kerem B.Rommens J,Buchanan J,Markewicz D,Cox T, et al.Identification of the cystic fibrosis gene:Genetics analysis.Science 1989;245:1074-1080


Más notas de la edición 49
Lee desde Issuu nuestra última edición publicada en Julio 2024, Edición número 155


Notas relacionadas a FIBROSIS QUÍSTICA Test del Sudor – Importancia...

