
Estudio descriptivo y de asociación con la esclerosis múltiple recurrente y su relación con la diversidad genética del complejo de receptores leucocitarios
Artículo extraído de la Memoria para optar al grado de Doctor presentada por David Ordóñez del Valle
Director: Carlos Vilches Ruiz
Universidad Complutense de Madrid, Facultad de Ciencias Biológicas.
Departamento de Biología Celular (Morfología Microscópica). Esta Tesis ha podido ser elaborada gracias a la financiación por parte de proyectos concedidos por la Fundación LAIR y el Ministerio de Educación y Ciencia (BFU2005–04622). Madrid, 2013
Correspondencia: Dr. David Ordóñez del Valle. OrdonezdelValle.David@mh-hannover.de
Klinik für Immunologie und Rheumatologie Medizinische
Hochschule Hannover Carl-Neuberg-Str. 1 30625 Hannover, Deutschland.
Tel. +49-(0)511-532-3626. Fax +49-(0)511-532-9067
Esclerosis Múltiple
La Esclerosis Múltiple es una enfermedad desmielinizante y neurodegenerativa del Sistema Nervioso Central (SNC). Es la principal causa de incapacidad crónica en adultos jóvenes, con una prevalencia de 40-60 casos por cada 100.000 habitantes en España [145]. Se caracteriza por el desarrollo de procesos in flamatorios del SNC mediados por células del Sistema Inmunológico, desmielinización y posterior degeneración axonal [146].
Formas clínicas y seguimiento de la Esclerosis Múltiple
En un 80-85% de los casos, la Esclerosis Múltiple se manifiesta con brotes puntuales y autolimitados de inflamación en el SNC (recurrencias) seguidos con fases de recuperación de los síntomas (remitencias), aunque con frecuencia tras la remitencia no se llega a recuperar totalmente el estado neurológico inicial y se va acumulando un cierto grado de incapacidad (Figura 1). Esta forma clínica se conoce como Esclerosis Múltiple Recurrente-Remitente (EM-RR). En algunos pacientes está descrita una evolución “benigna” a lo largo de los años sin incremento de la discapacidad mientras que en un 1-3% de los casos la Esclerosis Múltiple evoluciona de manera muy agresiva (Esclerosis Múltiple Maligna). Tras unos años de evolución, un 60% de los pacientes de RR-MS comienza a incrementar el grado de incapacidad de manera progresiva e independiente de los brotes inflamatorios, dando así lugar a otra forma clínica denominada Esclerosis Múltiple Secundaria Progresiva (EM-SP). Por tanto, a RR-MS y la SP-MS podrían considerarse como 2 fases de una misma enfermedad y en ocasiones pacientes diagnosticados de cualquiera de las dos formas clínicas se agrupan en una misma entidad que recibe el nombre de Esclerosis Múltiple Recurrente (EM-R) [146].
En un 10% de los pacientes de Esclerosis Múltiple existe otra evolución en la que el incremento del grado de discapacidad se produce de manera gradual ya desde las primeras manifestaciones de la enfermedad (Figura 1). Esta forma clínica recibe el nombre de Esclerosis Múltiple Primaria Progresiva (EM-PP) [146].

Figura 1 – Evolución del valor del EDSS en las distintas formas clínicas de la Esclerosis Múltiples.
Para determinar el estado puntual de afectación neurológica del paciente de Esclerosis Múltiple se ha desarrollado un parámetro denominado Escala Ampliada del Grado de Discapacidad (Expanded Disability Status Scale, EDSS – [147]) que varía en un rango de 0 a 10, siendo el 0 un valor de exploración neurológica normal y 10, fallecimiento. El parámetro MSSS (Multiple Sclerosis Severity Score) es un índice de la gravedad del curso clínico de la Esclerosis Múltiple que relaciona el grado de discapacidad (EDSS) con el tiempo que se ha tardado en alcanzarlo. El MSSS también se considera como un factor predictivo. Al igual que con el índice anterior, los valores del MSSS varían entre 0 y 10 [148].
Factores asociados al riesgo de desarrollar Esclerosis Múltiple
La Esclerosis Múltiple es una enfermedad de etiología desconocida, se considera que se produce por factores ambientales que afectan a individuos genéticamente predispuestos a desarrollarla. Están descritos varios factores ambientales y genéticos que se asocian a un mayor riesgo de desarrollar la Esclerosis Múltiple.
Factores ambientales
a) Latitud geográfica:
Aunque esta idea está en constante revisión, durante muchos años se consideró que la Esclerosis Múltiple presentaba una distribución geográfica con un gradiente Norte-Sur por el que la prevalencia de la enfermedad era más alta en regiones del Norte de Europa y América y disminuía gradualmente en países situados en latitudes inferiores. También se observaba un efecto migratorio según el que en territorios del hemisferio sur colonizados por Noreuropeos, como por ejemplo Australia o Nueva Zelanda, había una mayor prevalencia de la enfermedad [149].
b) Exposición solar – Vitamina D:
Ambos factores se encuentran muy relacionados entre sí y con la latitud geográfica, ya que la latitud influye directamente en la duración e intensidad de la exposición a la luz solar. La exposición a la radiación ultravioleta B (UV-B; 290-320nm) de la luz solar inicia el proceso de síntesis de la vitamina D [150]. Se ha observado que tanto la exposición solar prolongada como los niveles elevados de vitamina D en sangre se asocian a una mayor protección frente al desarrollo de la Esclerosis Múltiple [151-153].
c) Infección por el virus de Epstein Barr:
El virus de Epstein-Barr (EBV) es el agente que causa la mononucleosis infecciosa. Infecta a los linfocitos B y, una vez resuelta la infección, permanece en ellos de manera latente [154, 155]. La similitud en las epidemiologías de la Esclerosis Múltiple y la mononucleosis infecciosa propició la idea de la existencia de una posible relación entre ambas entidades [155]. La causa de esta asociación no está definida aún, aunque una posible explicación podría ser el mimetismo molecular entre antígenos del EBV y componentes de las vainas de mielina de los axones [156]. Así, están descritos linfocitos T CD4+ específicos frente a EBNA-1 (EBV encoded nuclear antigen) cuyos niveles están significativamente aumentados en pacientes de Esclerosis Múltiple [157-159] o la existencia de péptidos inmunorreactivos procedentes de proteínas del EBV en el líquido cefalorraquídeo de pacientes de Esclerosis Múltiple [160, 161]. El EBV también puede afectar a la función de los linfocitos B, ya que se han encontrado folículos linfoides secundarios ectópicos en meninges de algunos pacientes de SP-MS [162]. En resumen, se considera que la infección por el EBV en individuos genéticamente predispuestos puede ser un factor desencadenante de la enfermedad.
Otro factor determinante en el desarrollo de la Esclerosis Múltiple puede ser la edad a la que se produce la infección por el EBV. Así, la incidencia de la Esclerosis Múltiple se incrementa significativamente cuando se produce un contacto con el EBV a edades más tardías (mayor o igual a 20 años), mientras que si la infección tiene lugar en la infancia, la incidencia de la MS se reduce en un 50% [163]. Esta observación concuerda con la “hipótesis de la higiene” [152], según la cual un contacto más temprano con el agente infeccioso sería protector frente al desarrollo de la Esclerosis Múltiple.
Factores genéticos
a) Sistema HLA
En una región de un tamaño aproximado de 3’5 Mb en el brazo corto del cromosoma 6 (6p23.1) se encuentran los genes del complejo principal de histocompatibilidad humano, junto con otros genes relacionados con la respuesta inmunológica. Se distinguen los genes de las cadenas alfa de las moléculas de clase I (HLA-A, B, C, E, F y G) y los genes que codifican para las cadenas alfa y beta de las moléculas de clase II (HLA-DR, DQ, DP, DM y DO). La primera referencia de una asociación entre genes del HLA y Esclerosis Múltiple fue la existente con el haplotipo HLA-A*03 y B*07 [164]. La especificidad serológica DR2, que engloba a los alelos de clase II HLA-DRB1*15 y HLA-DRB1*16, se encuentra en desequilibrio de ligamiento con los anteriores y también se asociaba con un mayor riesgo de desarrollar la Esclerosis Múltiple [165, 166]. Actualmente se considera que el haplotipo de clase II que incluye los alelos DRB1*15:01-DRB5*01:01 – DQB1*06:02-DQA1*01:02 (también conocido como haplotipo DR15) es el principal marcador genético de riesgo [167-169]. El alelo HLA-DRB1*15:01 se encuentra significativamente aumentado en pacientes de Esclerosis Múltiple y su presencia aumenta 3.5 veces el riesgo de desarrollar la enfermedad en población sueca [170]. La mayor frecuencia de HLA-DRB1*15:01 se observó también en otras poblaciones [168] incluida la española (OR= 2.69 – 3.1) [171-173]. Además de este efecto directo, existen interacciones epistáticas entre HLA-DRB1*15:01 y otros genes de HLA de clase I y clase II [174].
Killer-cell Ig-like Receptors (KIR): En el LRC se encuentran los genes que codifican para los receptores KIR (CD158) en una región de ~150 kb del LRC conocida como el Complejo KIR, en una posición más telomérica respecto a los LILR (19q13.4). ]. Los KIR se expresan en la membrana de linfocitos con actividad citotóxica (células NK y Linfocitos T CD8+) [71] y algunos Linfocitos T CD4+ [72]. Algunos tienen como ligando las moléculas HLA de clase I, participando en la vigilancia de su expresión en las células del organismo [73, 74].
b) Otros genes implicados
En los Genome Wide Association Studies (GWAS) se han ido encontrando otros marcadores genéticos asociados a un mayor riesgo a desarrollar la Esclerosis Múltiple, como por ejemplo polimorfismos presentes en los genes de los receptores de las interleuquinas 2 y 7 (IL2RA – IL7R; OR= 1.3 – 1.24, respectivamente; [175]), o con un papel protector como es el caso de STAT3 (OR= 0.87; [176]) o CD58 (OR= 0.82; [177]). En la región 19q13 se han detectado marcadores de riesgo [178-180]. En el “Extended LRC” se describió la existencia de un microsatélite en el intrón 1 de ApoC2 (Apolipoproteína C2, 19q13.2) que incrementaba el riesgo de desarrollar Esclerosis Múltiple [181]. También en el “Extended LRC”, alelos de otras apolipoproteínas como el Epsilon4 de la ApoE se relacionan con un peor curso de la enfermedad [182, 183]. En una posición ~10 Mb más telomérica respecto a los genes ApoC2 y ApoE, se propuso que la deleción de LILRA3 (19q13.4) es un factor de riesgo genético asociado al desarrollo de la Esclerosis Múltiple en población alemana (OR= 2.2; [64]). LILRA3-del no ha sido analizado en ninguna otra población, incluyendo la española, en el contexto de la Esclerosis Múltiple y se desconoce si se trata de un factor de riesgo primario o asociado por desequilibrio de ligamiento a otros factores de riesgo localizados en el LRC, como por ejemplo la variabilidad KIR. Se desconoce, por otra parte, el posible efecto de las formas aberrantes de LILRA3 o de su variabilidad alélica en la etiopatogenia de la Esclerosis Múltiple. Asimismo, nunca se han analizado posibles interacciones epigenéticas de LILRA3-del con otros factores de riesgo presentes en otros cromosomas, principalmente HLA-DRB1*15:01.
Estructura y nomenclatura de los KIR
Los KIR son proteínas de membrana que pueden presentar 2 ó 3 dominios Ig-like extracelulares (KIR2D y KIR3D, respectivamente), una región transmembrana y una cola citoplásmica de longitud variable. Al igual que en los LILR, los KIR inhibidores tienen una cola citoplásmica larga (long – L) con 2 motivos ITIM y los KIR activadores tienen una cola citoplásmica corta (short – S) que carece de motivos señalizadores, pero tienen en la región transmembrana un residuo de lisina que les permite su asociación con la molécula activadora DAP12 (Figura 2 ver a la derecha). El número final en el nombre de un KIR se refiere a loci o grupos alélicos distintos de receptores KIR con estructuras similares. De esta manera, KIR3DL1 es uno de los receptores descritos con 3 dominios Ig-like extracelulares y una cola larga que envía señales de inhibición [75].
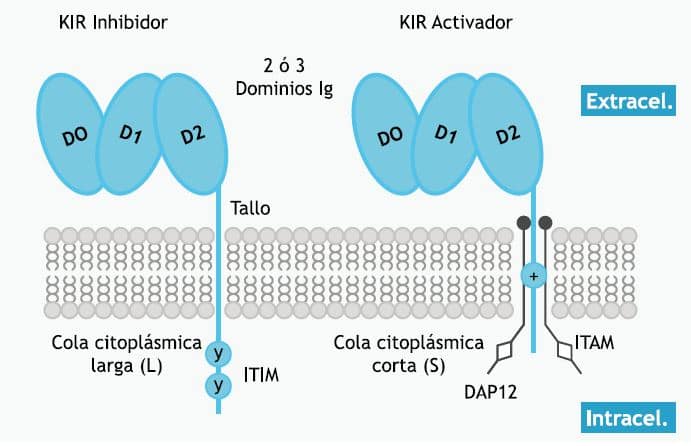
Figura 2 – Estructura de los receptores KIR inhibidores y activadores

Función de los KIR
Como resultado de la interacción KIR-HLA, la célula citotóxica puede recibir señales activadoras o inhibidoras según el receptor que está participando. En este sentido, esta bien descrito el papel que desempeñan los receptores KIR inhibidores, ajustándose a la hipótesis del “missing self” [76]. Los linfocitos T CD8+ detectan la presencia de células neoplásicas o infectadas por virus reconociendo péptidos extraños presentados en HLA clase I. Como resultado, se produce la activación del linfocito T liberando el contenido de sus gránulos de granzima y perforina para lisar a la célula diana (Figura 3). En células tumorales o infectadas por virus, la presentación antigénica puede verse alterada al bloquearse la expresión en membrana de moléculas HLA de clase I. Por ejemplo, el Citomegalovirus humano (hCMV) presenta varios mecanismos que alteran la expresión en membrana de la molécula HLA de clase I [77, 78]:
- las proteínas US2 y US11 se expresan en el retículo endoplásmico (RE) e inducen la degradación de la cadena a del HLA de clase I por el proteasoma;
- la proteína US6 impide la carga del péptido en el HLA bloqueando el transporte de péptidos desde el citoplasma al interior el RE a través de TAP;
- US3 y US10 bloquean el transporte de la molécula HLA de clase I con el péptido cargado desde el RE al aparato de Golgi, quedando retenida en el RE. La proteína E19 del adenovirus retiene la molécula HLA en el RE interfiriendo en la asociación de la molécula HLA con la tapasina, o induciendo su transporte de vuelta del aparato de Golgi al RE [79, 80]. El virus del herpes simplex tipo I también bloquea el paso de péptidos a la luz del RE a través de TAP mediante la proteína ICP47 [81, 82]. Otro ejemplo es el herpesvirus asociado al Sarcoma de Kaposi, que codifica para dos proteínas K3 y K5 que disminuyen la expresión de membrana del HLA induciendo procesos de endocitosis por vesículas revestidas de clatrina [83, 84].
Estas alteraciones de la expresión antigénica evita la detección y posterior lisis de células infectadas o tumorales por parte de los linfocitos T citotóxicos. Cuando una célula NK reconoce, mediante un KIR inhibidor, la presencia de moléculas HLA de clase I en la membrana de células sanas se frena la respuesta citotóxica. Por el contrario, si la expresión de las moléculas HLA se ve disminuida o bloqueada (Figura 3), la señalización por los KIR inhibidores se ve afectada al no reconocer la expresión del ligando en la célula alterada y ésta sería lisada por la célula NK [85-87], si además presenta estructuras reconocidas por los receptores activadores.

Figura 3 – Comparación de las lisis de células diana mediada por células citotóxicas al reconocer la expresión del ligando en el caso del Receptor de Linfoncitos T (T Cell Receptor – TCR, izq.) o por la ausencia del ligando en el caso de los KIR según la hipótesis del “missing self” (der). La expresión de receptores activadores de células NK se ha omitido para simplicar la figura.
El papel activador de los KIR asociados a la molécula DAP12 no encaja en la hipótesis del “missing self” aunque se cree que pueden actuar como moléculas coestimuladoras de células citotóxicas en presencia de su ligando en la célula diana. Así, KIR2DS1 reconoce la expresión de HLA-C2, aunque con menor afinidad que su homólogo inhibidor KIR2DL1, y es capaz de activar por sí sólo a células NK para lisar células transformadas con el EBV, o Linfoblastos T y células dendríticas alogénicas que presentan su ligando [88]. KIR2DS4 reconoce diferentes moléculas HLA-C y también a HLA-A*11, y la unión con este último es capaz de activar a las células NK en ensayos de citotoxicidad [89]. KIR2DS2, KIR2DS3, KIR2DS5 y KIR3DS1 no tienen ligando descrito, aunque este último podría reconocer a HLA-Bw4, al igual que su homólogo inhibidor KIR3DL1 [90].
Haplotipos KIR
El “haplotipo KIR” es la disposición de los genes KIR en cada cromosoma 19. Una característica diferencial de los haplotipos KIR es la variación en el contenido génico. De esta manera, existen unos genes que están presentes en el ~100% de los haplotipos KIR descritos y que se conocen como “framework genes”. KIR3DL3 y KIR3DL2 delimitan respectivamente, los extremos
centromérico y telomérico del haplotipo KIR, mientras que los loci KIR3DP1 y KIR2DL4 conforman la zona central. Estos genes definen las regiones centroméricas y telómericas y dentro de ellas hay una gran variabilidad entre haplotipos por los genes que están presentes (Figura 4 ver a la derecha) [116]. Esta es una diferencia importante con los genes del Complejo LILR, en el que no hay variación en el número de copias de los genes presentes con la excepción de LILRA3.

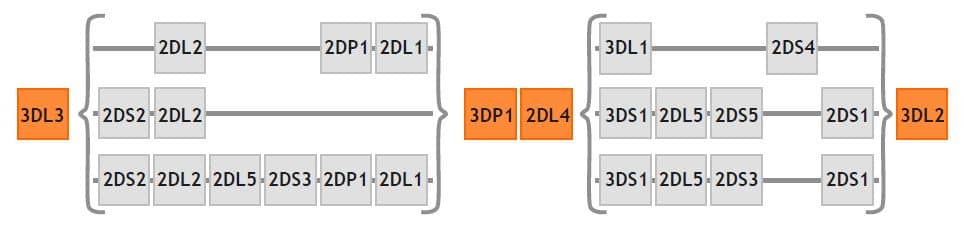
Figura 4 – Genes KIR – Haplotipos
Se distinguen dos grupos de haplotipos KIR. El haplotipo A tiene una dotación génica constante que incluye los loci KIR3DL3, KIR2DL3, KIR2DP1, KIR2DL1, KIR3DP1, KIR2DL4, KIR3DL1, KIR2DS4 y KIR3DL2. Este haplotipo es el mayoritario y solo porta un gen que codifica para un receptor activador (KIR2DS4) que, en un 75% de la población Caucasoide, está representado por alelos que tienen una deleción de 22pb en el exón 5 (D2) y generan una forma corta de KIR2DS4 de significado fisiológico desconocido [117]. Por tanto, en la mayoría de los casos el haplotipo A solo porta receptores inhibidores de la respuesta citotóxica. La variabilidad entre distintos haplotipos A se consigue gracias a diferentes combinaciones de polimorfismos alélicos de los loci que incluye.
Cualquier variación por exceso respecto a esta dotación génica de los haplotipos A se considera un haplotipo B. En el haplotipo B hay una mayor heterogeneidad génica y puede incluir regiones centroméricas y teloméricas de haplotipos A. Esto se debe a que muchos de los haplotipos B se generan por procesos de recombinación a nivel de la región intergénica entre KIR3DP1 y KIR2DL4. En los haplotipos B hay una mayor presencia de receptores activadores, con una importante variabilidad en el número de genes de estos receptores que pueden estar representados en los distintos haplotipos.
Esclerosis Múltiple y Células NK
Las células NK ejercen una doble función, por un lado son capaces de lisar células infectadas o con transformaciones neoplásicas y también pueden secretar citoquinas como IFN-gama. Se distinguen las células NK CD56bright (de alta expresión del marcador CD56; neural cell adhesion molecule – NCAM) KIR- que al activarse proliferan y se encargan de la secreción de citoquinas; y las CD56dim (de baja expresión de CD56) KIR+ y tienen capacidad citolítica. Estas 2 poblaciones de Células NK están muy relacionadas entre sí y se considera a las CD56bright como posibles precursoras de las CD56dim [184].
El papel de las células NK en la patogenia de la Esclerosis Múltiple aún no está definido. Las células CD56bright parecen ejercer un papel beneficioso en la Esclerosis Múltiple al aumentar sus niveles en respuesta a los diferentes tratamientos como daclizumab [185], interferon-beta [186] y ciclofosfamida [187]. Por otra parte, las primeras referenciasde estudios de células NK en la Esclerosis Múltiple reflejaron que la disminución de la actividad citolítica de las células NK se correlacionaba con las exacerbaciones de la sintomatología mientras que en las remitencias, las células NK recuperaban su actividad [188-190]. Por otra parte se ha observado una capacidad citolítica de las células NK frente a oligodendrocitos y otras poblaciones de células gliales (astrocitos y microglía) mediada por el receptor NKG2D [191].

Los KIR participan en la defensa frente a infecciones causadas por Herpesvirus [78, 129], que pueden ser el factor desencadenante de desarrollar la Esclerosis Múltiple por su asociación con el EBV, y los niveles de expresión de los KIR en Linfocitos T o en células NK se ven afectados por el curso clínico y por tratamientos como el IFN-beta [192, 193]. La gran variabilidad existente en el Complejo KIR nunca ha sido analizada en el contexto de la Esclerosis Múltiple y, por la proximidad del Complejo KIR a un posible factor de riesgo como LILRA3-del, puede existir una asociación primaria entre los KIR y la Esclerosis Múltiple o por desequilibrio de ligamiento de los KIR y LILRA3-del.
Los LILR (CD85) son una familia de receptores expresados por todas células del Sistema Inmunológico y reconocen moléculas HLA de clase I
LILRA3 es una proteína de 439 aminoácidos (~47.5 kDa) secretada principalmente por monocitos y se detecta en suero (~2700 pg/mL) y en líquido sinovial de pacientes con Artritis Reumatoide [57]. Recientemente se ha descrito su capacidad para unirse a HLA-A*02:01, HLA-G1 [58] y a la cadena alfa libre de la molécula HLA-C [59], aunque se desconoce el efecto fisiológico. La isoforma corta de LILRA3 daría lugar a un receptor secretado de menor tamaño (375 aminoácidos) y peso (40.3 kDa); desconociéndose también su implicación fisiológica. En la población sana está descrita la presencia de una forma alterada de LILRA3 por una deleción de 6765 pb que recibió el nombre no oficial LILRA3*005 [44, 60], aunque a partir de ahora nos referiremos a ella como LILRA3-del (Figura 5). La deleción comienza 3.5 Kb más arriba del codón de inicio de LILRA3, incluye a la región promotora y a los exones que codifican para el péptido señal y los cuatro dominios Ig-like, y finaliza en la región 3’ del intrón 6, a 752 pb del exón 7. Esta deleción, por tanto, afecta a la práctica totalidad de la secuencia codificante en LILRA3. Su frecuencia es de un 20-30% de la población sana Caucasoide [63-65] con una frecuencia alélica de 0.26 en población británica [60] y puede llegar hasta un 70-85% de los individuos de poblaciones de raza Mongoloide [61, 66].

Figura 5 – Esquema de la deleción que afecta a la práctica totalidad de la secuencia codificante de LILRA3
Esta alteración en la secuencia de LILRA3 puede tener una implicación funcional sobre la regulación del Sistema Inmunológico, ya que existen trabajos publicados que detectan un aumento de la frecuencia de LILRA3-del en pacientes de enfermedades de base autoinmune como la Esclerosis Múltiple [64] o el Síndrome de Sjögren [65]. Por el contrario, no se ha encontrado asociación entre la presencia de la deleción y un mayor riesgo de desarrollar psoriasis [63].
Esclerosis Múltiple y diversidad en el Complejo de Receptores Leucocitarios (LRC)
Variabilidad en el Complejo KIR La variabilidad genética en el Complejo KIR ha demostrado su implicación en enfermedades de base autoinmune como la psoriasis [206], diabetes mellitus [207] esclerodermia [135, 208]. La EM-R es una enfermedad autoinmune de carácter multifactorial con un importante componente genético. Se han encontrado polimorfismos de susceptibilidad en el cromosoma 19 [209] y también está descrita la implicación de la apolipoproteína E (ApoE, 19q13.2) como posible factor asociado con un peor pronóstico de la EM-R (revisado por Pinholt et al, [210]). En una región relativamente cercana, 19q13.4, se había descrito la deleción de LILRA3 como otro posible factor que predispone a desarrollar la EM-R en población alemana aunque sin precisar si se trata de un factor primario o secundario [64].

La variabilidad del Complejo KIR, por el contrario, nunca había sido analizada en la Esclerosis Múltiple. Encontramos un posible papel protector de KIR3DS1, que es un gen incluido en la región telomérica de los haplotipos B KIR3DS1-KIR2DL5A-KIR2DS3S5-KIR2DS1. En estudios posteriores, esta región (concretamente la presencia de KIR2DS1) también manifestó un efecto protector frente a la MS en población italiana [211]. KIR3DS1 es un receptor activador que interacciona epistáticamente con HLA-Bw4, ligando de su homólogo inhibidor KIR3DL1, observándose una progresión más lenta del SIDA [212]. La presencia de alelos del grupo HLA-Bw4 parece proteger frente al desarrollo de la Esclerosis Múltiple [213], aunque resulta contradictorio que el efecto combinado de la presencia de KIR3DS1 y HLA-Bw4 haya resultado en un aumento del riesgo en otro estudio en población española [214]. Todos los análisis efectuados hasta la fecha se resumen en la Tabla 1, sin demostrar ningún efecto concluyente de la implicación de la variabilidad KIR en la EM-R, por lo que serán necesarios análisis en series más largas para confirmar el papel real de la variabilidad KIR en la Esclerosis Múltiple.
Tabla 1 (ver a la derecha) – Resumen de los estudios de asociación de la variabilidad del Complejo KIR con el desarrollo de la MS en el que se analiza la reproducibilidad de nuestro resultado observado en KIR3DS1 y KIR2DS1. Los valores de la Odds Ratio (OR) en negrita son aquellos que alcanzan la significación estadística. Valores de significación no corregidos por el test de Bonferroni.

(1) El resto de trabajos están realizados en individuos caucásicos – (2) Trabajo presentado en el KIR Workshop de Estocolmo, Junio 2011
Esclerosis Múltiple y LILRA3:
Deleción de LILRA3
En enfermos de EM-R españoles replicamos el resultado obtenido en población alemana que asociaba la deleción de LILRA3 con el incremento del riesgo a desarrollar la enfermedad. Además, escribimos por primera vez que este aumento sigue una tendencia lineal por la que el riesgo se incrementa de manera significativa al estar presente la deleción de LILRA3 en homocigosis. Este resultado también se observaba en el estudio realizado en población alemana, aunque no había sido comentado por los autores [64].
En otro análisis de la deleción en homocigosis realizado en población finlandesa se observó una mayor frecuencia en pacientes con EM-R, aunque no llegaba a ser significativa [209]. No existen más trabajos que analicen la frecuencia de la deleción de LILRA3 en laEsclerosis Múltiple, pero sí se analiza en otra patologías como la Psoriasis [63], la enfermedad celiaca [216] o el Síndrome de Sjögren [65] y sólo en la última se observaron diferencias significativas entre pacientes y controles. Si comparamos la frecuencia de la deleción de LILRA3 en población española con la del resto de poblaciones europeas se observa un gradiente Norte-Sur, siendo mínima su frecuencia en España (Figura 6). No existen más datos de distribución de la deleción en países de la cuenca mediterránea, con la excepción de un análisis realizado en Palestina que presenta unos valores similares a los de España [62]. Este gradiente Norte-Sur observado en la distribución de LILRA3-del, se asemeja al descrito en la epidemiología de la Esclerosis Múltiple [149], lo que podría concordar con una posible implicación de LILRA3 en la EM-R.

Figura 6 – Distribución de las frecuencias alélicas de la deleción de LILRA3 en Finlandia, Alemania ([64, 65]), Reino Unido ([60]), Polonia ([63]) y Palestina ([62]). Las frecuencias en España proceden de nuestro trabajo y el valor en Finlandia no está publicado, proviene de una referencia personal del autor (Dr. PJ Tienari; Helsinki University Central Hospital, Haartmaninkatu, 4, Helsinki, Finlandia). En fondo azul, los valores de frecuencia de la deleción en enfermos de EM-R
Variabilidad alélica de LILRA3
Las formas alteradas de LILRA3 (alelos LILRA3*011N y *012N) descritas en población asiática [61] nunca habían sido analizadas en los estudios de asociación de LILRA3 con las enfermedades autoinmunes y se incluían dentro de la forma no delecionada de LILRA3, a pesar de que sus alteraciones en la secuencia pueden afectar completamente a la funcionalidad del receptor. Estos alelos son relativamente comunes en poblaciones del Este de Asia y se desconocía su frecuencia en población Caucasoide, con la excepción de una pequeña serie de 59 controles sanos de la Foundation Jean Dausset – Centre d’Etude du Polymorphisme Humain (CEPH, Paris) y en la que en ninguno de ellos fue detectado [66]. Dado que la ausencia de un receptor LILRA3 funcional es un factor de riesgo para el desarrollo de la EM-R, analizamos la presencia de estos alelos aberrantes de LILRA3 en nuestras series de controles y pacientes (519 individuos en total) y solo encontramos un paciente LILRA3+/del con una de las formas no funcionales (LILRA3*011N). Este resultado indica que los alelos LILRA3*011N y *012N son muy infrecuentes en la población española y dificulta su evaluación como posible factor de riesgo. En cualquier caso, la hipotética implicación de estos alelos en la etiología de la EM-R en España sería mínima por su baja frecuencia.
También analizamos las variantes alélicas posiblemente funcionales de LILRA3 en ambas series. Su distribución en controles sanos de origen caucasoide ya se había realizado en población del Reino Unido [60] y nuestra serie es la primera referencia en población europea del área mediterránea. No encontramos diferencias significativas entre las distribuciones alélicas y genotípicas de nuestras series de pacientes con EM-R y controles, ni tampoco observamos una aceleración del curso clínico de la enfermedad. En cambio, sí detectamos un posible efecto aditivo con la presencia de la deleción incrementando el riesgo a desarrollar la EM-R. Este resultado se ajusta la hipótesis por la que la ausencia de alteraciones o de variabilidad en el receptor LILRA3 es un factor protector al desarrollo de la R-MS.
Deleción de LILRA3 y la variabilidad en el Complejo KIR en la EM-R
Los Complejos LILR y KIR se encuentran contiguos en el LRC, lo que nos llevó a evaluar la posible existencia de un desequilibrio de ligamiento entre ellos que explicase la desviación observada en la frecuencia de la deleción en pacientes con EM-R. Nuestros datos concuerdan con un trabajo previo en el que no se observa un desequilibrio de ligamiento significativo entre la LILRA3-del y ninguno de los genes KIR [62], por lo que el Complejo KIR no determina la asociación de la deleción con la EM-R. El locus más centromérico del Complejo KIR es KIR3DL3 y su intrón 5 es un punto caliente de recombinaciones [118, 217]. Esta variabilidad junto con la distancia cromosómica entre LILRA3 y KIR3DL3 (~400 kb) pueden ser las causas de la ausencia desequilibrio de ligamiento entre el Complejo KIR y LILRA3-del.
Posible papel de LILRA3 en la EM-R
La EM-R es una enfermedad multifactorial en la que se produce una desregulación del Sistema Inmunológico y en la que la ausencia de LILRA3 o una disminución de sus niveles podrían estar implicadas. LILRA3 parece tener un efecto anti-inflamatorio, en contraposición a otros receptores activadores como LILRA1 y LILRA2, dado que su secreción “in vitro” por monocitos se ve favorecida por la presencia de citoquinas anti-inflamatorias (IL-10) o por la privación de factores pro-inflamatorios como el TNF alfa [57]. Respecto a la EM-R, en las recurrencias tienen lugar procesos inflamatorios que se asocian a reacciones autoinmunes sobre las vainas de mielina que acaban provocando daños axonales en el SNC. La función de LILRA3 en este contexto podría ser la de regular la inflamación en el SNC y su ausencia o la disminución de sus niveles podrían estar implicadas tanto en el origen e intensidad de los procesos inflamatorios como en su resolución. De nuestros datos no se puede extraer estas conclusiones, aunque si observamos que el curso clínico de la enfermedad se acelera por la presencia conjunta del alelo HLA-DRB1*15:01 y de la deleción de LILRA3 (Figura 7).
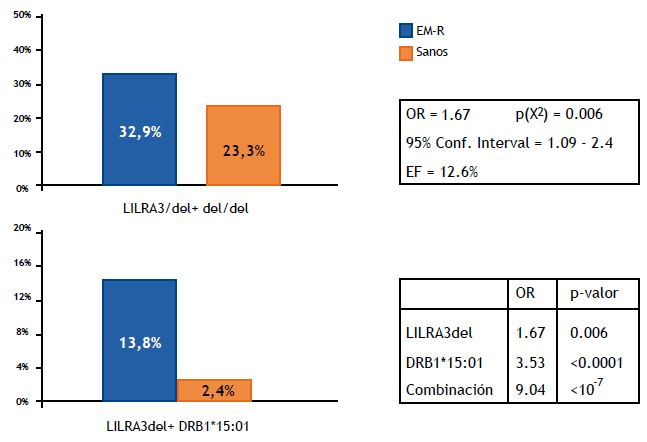
Figura 7 – Arriba, frecuencia de la presencia de la delecion de LILRA3 en pacientes y controles. Abajo a la izquierda, presencia conjunta de la deleción y el alelo HLA-DRB1*15:01 en pacientes y controles. Abajo a la derecha, comparación de las Odds ratio (OR) de cada factor por separado con la OR de la presencia conjunta de ambos factores.”
Se ha descrito la presencia de LILRA3 de manera constitutiva en el suero de controles sanos y sus niveles se ven aumentados en pacientes de Artritis Reumatoide, una enfermedad de base autoinmune. En estos pacientes, LILRA3 también se detecta en muestras de líquido sinovial procedente de las lesiones inflamatorias [57]. Aún no ha sido analizada la posible presencia de LILRA3 en muestras de líquido cefalo-raquídeo de pacientes con EM-R, ya sea de manera constitutiva o debida a la enfermedad. Por otra parte, en el tratamiento con IL-10 frente a la psoriasis se incrementan de manera significativa los niveles de ARNm de LILRA3 en monocitos [218], resultados reproducidos “in vitro” al analizar PBL’s procedentes de pacientes de Artritis Reumatoide [57].
Dado que los monocitos producen mayor cantidad de LILRA3 en presencia de estímulos anti-inflamatorios, en pacientes con genotipos LILRA3+/+ o LILRA3+/- se podría realizar una estimulación de monocitos “ex-vivo” y volver a administrarle estos monocitos activados. Otra aproximación sería diseñar fármacos que estimulen a los monocitos de manera específica, como se pudo observar en el caso de la administración de IL-10, sin necesidad de realizar tratamientos “in vitro”. Debido a que el efecto de la administración de IL-10 es de acción inespecífica, ya que estimula o inhibe a otros genes, lo ideal sería diseñar fármacos que incrementen la producción de ARNm de LILRA3 de la forma más específica posible.
Si se confirma que la alteración de LILRA3, ya sea por la completa inactivación de su expresión o por modificaciones de su funcionalidad, está implicada en la patogenia de la EM-R o de otras enfermedades autoinmunes se podría tratar de restaurar los niveles del receptor en pacientes LILRA3+/- mediante la administración de LILRA3 producido “in vitro”. Esta aproximación supondría un mayor riesgo en el caso de pacientes LILRA3-/- debido a la posibilidad de desarrollar reacciones de hipersensibilidad ya que su sistema inmunológico no estaría tolerizado frente a LILRA3. Una última posibilidad en estos pacientes LILRA3-/-, aunque con más dificultades técnicas, sería intentar reconstituir la expresión de LILRA3 mediante terapia génica sobre sus propias células progenitoras del Sistema Inmunológico.
Conclusiones
- La deleción de LILRA3 se asocia al desarrollo de la EM-R en España e interacciona con HLA-DRB1*15:01 incrementando el riesgo de desarrollar la enfermedad y, posiblemente, acelerando su curso clínico.
- Los alelos aberrantes de LILRA3 son muy infrecuentes en España, lo que impide evaluarlos como posible factor de riesgo.
- Los polimorfismos de LILRA3 no incrementan significativamente el riesgo a desarrollar la EM-R ni empeoran su curso clínico, aunque si observamos un posible efecto aditivo sobre el riesgo conferido por la deleción.
- No existen interacciones consistentes entre los polimorfismos de LILRA3 y el alelo HLA-DRB1*15:01.
- El haplotipo telomérico B, compuesto por KIR3DS1-KIR2DL5A-KIR2DS3S5-KIR2DS1, podría tener un efecto protector frente a la EM-R

Más notas de la edición 30
Lee desde Issuu nuestra última edición publicada en Octubre 2025, Edición número 170

Notas relacionadas a Estudio descriptivo y de asociación con la...

