

Enfermedad de Addison como presentación de un síndrome pluriglandular autoinmune tipo 2
Los síndromes plurigandulares autoinmunes se definen por la coexistencia de al menos dos insuficiencias glandulares a consecuencia de una pérdida de la inmunotolerancia. Existen 4 tipos, siendo el tipo 2 el más frecuente.
Casuística de interés
Dr. Santiago Acle1 ; Dra. Ximena Añón2 ; Dr. Álvaro Danza3 ; Dr. Raúl Pisabarro4
1Residente Clínica de Médica “A”. Facultad de Medicina. UdelaR. Montevideo.2Residente Clínica de Médica “A”. Facultad de Medicina. UdelaR. Montevideo.3Asistente Clínica de Médica “A” y del Departamento de Farmacología y Terapéutica. Facultad de Medicina. UdelaR. Montevideo.4Profesor Agregado de Clínica Médica “A”. Facultad de Medicina. UdelaR. Montevideo.Arch. Med Int v.33 n.3 Montevideo dic. 2011 Trabajo de la Clínica Médica “A”. Departamento Clínico de Medicina. Hospital de Clínicas “Dr. Manuel Quintela”. Facultad de Medicina. UdelaR. Correspondencia: Dr. Santiago Acle, Vásquez Ledesma 2969 Apartamento 901. Tels.: 6138056 – 099929346. Email:santiagoacle@gmail.com
Resumen.
Los síndromes plurigandulares autoinmunes se definen por la coexistencia de al menos dos insuficiencias glandulares a consecuencia de una pérdida de la inmunotolerancia. Existen 4 tipos, siendo el tipo 2 el más frecuente. Es condición indispensable la presencia de insuficiencia suprarrenal primaria para hacer el diagnóstico de éste último, pudiendo muchas veces ésta preceder a las otras endocrinopatías que lo conforman: enfermedad tiroidea autoinmune y/o diabetes mellitus tipo 1. La insuficiencia suprarrenal primaria se caracteriza por la producción adrenal deficiente de cortisol, mineralocorticoides y andrógenos. Es una enfermedad poco frecuente que se presenta habitualmente en mujeres en edad media, siendo su etiología más prevalente la adrenalitis autoinmune. Se discute el caso de una paciente de 35 años que consulta por híperpigmentación de piel y episodios presincopales como presentación de una Enfermedad de Addison.La paciente presentó además amenorrea de 8 años de evolución y un bocio grado II lo cual nos llevó a plantear que era portadora de un síndrome plurigandular autoinmune tipo 2.
Palabras clave: Síndrome pluriglandular autoinmune, insuficiencia suprarrenal primaria, enfermedad tiroidea autoinmune, diabetes mellitus tipo 2.
Summary: Arch Med Interna 2011 – XXXIII(3):65-69.
Polyglandular autoimmune syndromes are defined by the coexistence of at least two glandular deficiencies resulting from the loss of immune tolerance. There are 4 types, being type 2 the most frequently seen. The presence of primary adrenal insufficiency is essential to make diagnosis of type 2 polyglandular autoimmune syndrome, and it may precede the other two endocrinopathies that compose it: autoimmune thyroid disease and/or diabetes mellitus type 1. Primary adrenal insufficiency is characterized by deficient production of adrenal cortisol, mineralocorticoids and androgens. It is a rare disease that usually occurs in middle-aged women being autoimmune adrenalitis its most prevalent etiology. In this article, we discuss the case of a 35 year old patient who complained of skin hyperpigmentation and presyncopal episodes as presentation of an Addison disease. The patient also presented amenorrhea of 8 years of evolution and a grade II goiter which led us to the diagnosis of a polyglandular autoimmune syndrome type 2.
Keywords: Autoimmune polyglandular syndromes, primary adrenal insufficiency autoimmune thyroid diseases, type 1 diabetes mellitus.
Introducción
Los síndromes pluriglandulares autoinmunes (SPA) se definen por la presencia de dos o más insuficiencias glandulares endócrinas como consecuencia de un trastorno inmunomediado. En 1980 Neufeld y Blizzard introdujeron este término en la literatura médica para describir la asociación entre insuficiencia suprarrenal primaria (ISP) de causa autoinmune, tiroidopatía autoinmune y candidiasis crónica cutáneomucosa (1). Apartir de ese momento se delimitaron dos tipos principales de SPA: el tipo 1, dado por la asociación de al menos dos de las siguientes condiciones: ISP, candidiasis crónica o hipoparatiroidismo crónico y el tipo 2, que asocia ISP, enfermedad tiroidea autoinmune y/o diabetes mellitus (DM) tipo 1 (2). Actualmente hay descriptas otras asociaciones menos frecuentes (insuficiencia gonadal primaria, hipopituitarismo, etc.) además de la presentación conjunta con otras enfermedades de naturaleza autoinmune no endocrinológicas, como vitíligo, anemia perniciosa, enfermedad celíaca y hepatitis. Al momento existen 4 tipos de SPA, siendo el SPA tipo 2 el más frecuente(1-3).
La ISP o enfermedad de Addison es una patología infrecuente en la población general, presentándose con una sintomatología poco específica en sus etapas iniciales, lo que determina frecuentemente un retraso en su diagnóstico. Debe ser tenida en cuenta como diagnóstico diferencial en consultas por pérdida de peso, fatiga e hipotensión, ya que un tratamiento oportuno puede mejorar significativamente la sintomatología, complicaciones y el pronóstico del paciente (4,5). Presentamos el caso de una paciente que consulta por híperpigmentación, alopecia y episodios presincopales como presentación de una enfermedad de Addison en el contexto de un SPA.
Caso Clínico
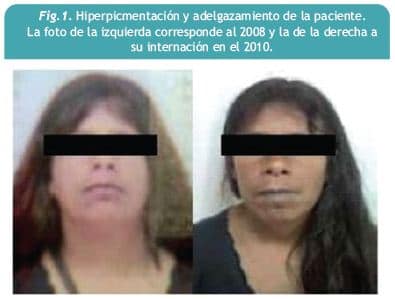
Mujer de 35 años, raza blanca, que comienza un año previo a la consulta con híperpigmentación de piel a nivel de cara y pliegues, comprometiendo progresivamente todo el cuerpo, mucosa de labios y faneras. El cuadro se acompaña de alopecía de axilas y miembros inferiores y a delgazamiento de aproximadamente 15 kg. El mes previo a la consulta presenta reiterados episodios presincopales. No relató fiebre ni síntomas respiratorios. Antecedentes personales: tabaquista, no tuberculosis, negaba conductas de riesgo para enfermedades de trasmisión sexual. Antecedentes ginecoobstétricos: amenorrea desde hace 8 años. Antecedentes familiares: desconoce madre y padre, 3 hijos sanos. En la exploración física al ingreso se constata: regular estado general, desnutrición proteico 2 calórica (IMC de 18 kg/m ), presión arterial de 100/50 mmhg, híperpigmentación de piel y mucosas, melanoniquia de uñas de manos y pies, bocio grado II y a nivel cardiovascular un ritmo regular de 54 cpm junto a hipotensión ortoestática (Figuras 1 y 2).
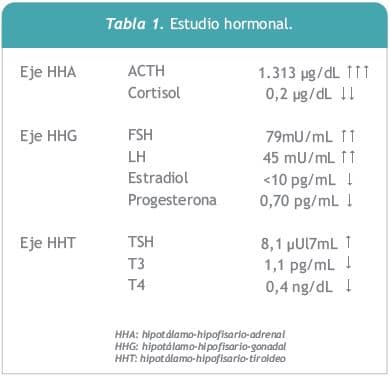
De la analítica básica de laboratorio se destaca: Na+ : 124 mmol/Ly K+ : 5,6 mmol/L, hemograma, azoemia, creatinina, funcional y enzimograma hepático: sin alteraciones. En lo que refiere al estudio hormonal se evidenciaron los siguientes resultados: ACTH francamente elevada: 1.323 ?g/mL, (VN: 7 – 63,3) y cortisol muy por debajo del límite normal: 0,2 ?g/dL (VN: 6 -19); FSH y LH ambas elevadas; estradiol y progesterona muy disminuidas; TSH: duplicando su valor normal y T3 y T4 libre descendidas. (Tabla I). La determinación de la actividad renina plasmática y la dosificación de aldosterona resultaron normales. El estudio inmunológico mostró: anticuerpos antiadrenales, antitiroglobulina y antiperoxidasa positivos, mientras que los anti-transglutaminasa tisular y los anti-gladina, buscando enfermedad celíaca, fueron negativos. A nivel imagenológico, la tomografía de abdomen y pelvis mostró las glándulas suprarrenales de tamaño normal sin evidencias de calcificaciones, la ecografía de tiroidesconfirmó la presencia de bocio difuso no nodular y la radiografía de tórax fue normal.
A la vista del cuadro clínico y los resultados paraclínicos analizados se hizo diagnóstico de ISP o enfermedad de Addison, asociada a un hipotiroidismo primario de causa autoinmune bajo la forma de tiroiditis de Hashimoto. Estas dos entidades conforman el SPAtipo 2, que en el caso de la paciente que se presenta se asoció a un hipogonadismo hipergonadotrópico (insuficiencia ovárica prematura) de probable etiología autoinmune, como manifestación de una enfermedad endocrinológica menor.
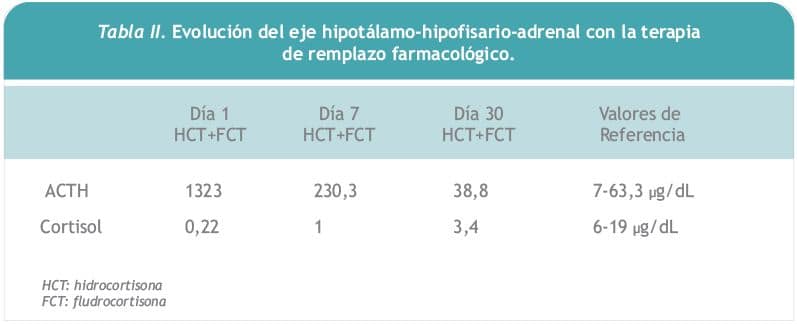
La paciente recibió como tratamiento dieta con balance adecuado de sodio, terapia sustitutiva en base a hidrocortisona 30 mg/día y fludrocortisona 0,1 mg/día, complementando en la evolución con levotiroxina 50 ?gs/día y terapia de remplazo hormonal con estrógenos y progestágenos. Presentó una notable mejoría de los síntomas, con un rápido asenso ponderal, desaparición de los episodios de hipotensión y normalización de las alteraciones hidroelectrolíticas. Al mes de iniciado la terapia de remplazo con glucocorticoides y mineralocorticoides los valores de ACTH se normalizaron y las concentraciones de cortisol presentaron un significativo ascenso (Tabla II).
En controles posteriores se le realizó una densitometría ósea, estudio del metabolismo fosfocálcico y dosificación de 250 de vitamina D, los cuales fueron normales, y se insistió en el estudio de sus hijos, dado que el SPAes una enfermedad de carácter hereditario.
Discusión
Los SPAse caracterizan por la coexistencia de al menos dos insuficiencias glandulares a consecuencia de una pérdida de la inmunotolerancia por autoanticuerpos o linfocitos T activados frente a antígenos propios (2). En su etiopatogenia interviene un infiltrado inflamatorio crónico a predominio linfocitario que genera una lenta y progresiva destrucción de los órganos endócrinos. Esta gradual evolución ocasiona un periodo preclínico donde son detectables los anticuerpos frente a la célula diana sin que haya una destrucción total, y un periodo posterior que, dejado a su libre evolución puede llevar a la insuficiencia glandular. En el año 2002 Bettelle et al. publicaron una clasificación basada en la propuesta por Neufeld y Blizzard que divide a los SPA en 4 tipos: SPA tipo 1: debe presentar al menos 2 de las siguientes características: candidiasis crónica, hipoparatiroidismo crónico e ISP; SPA tipo 2 o síndrome de Schmidt: obligatoriamente cursa con ISP asociando a enfermedad tiroidea autoinmune y/o DM tipo 1; SPA tipo 3: presenta enfermedad tiroidea autoinmune y otras enfermedades autoinmunes, exceptuando la ISP, la candidiasis crónica o el hipoparatiroidismo; SPA tipo 4: 2 o más enfermedades autoinmunes específicas de órgano que no cumpla los criterios de tipo 1, 2 ni 3 (2)(Tabla III).

SPA tipo 2 o ex sÍndrome de Schmidt
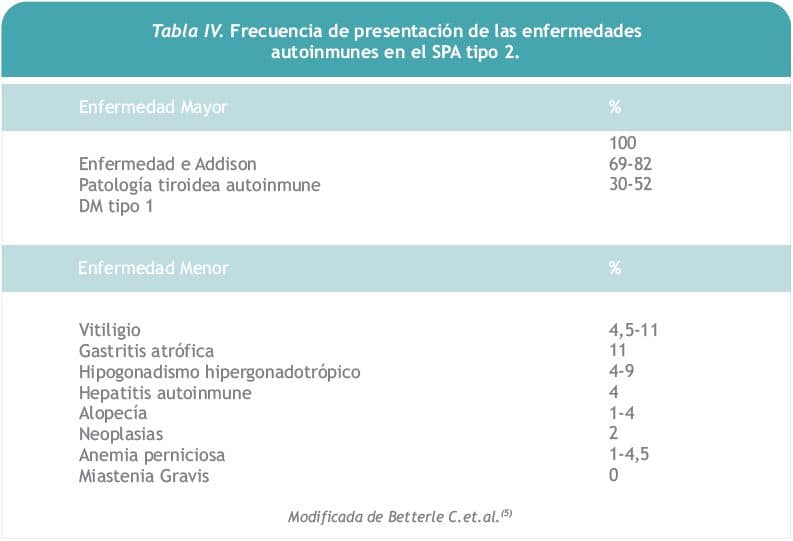
Su prevalencia se ha estimado en 1,5 a 4,5 casos por 100.000 habitantes, siendo el más común de los SPA. Afecta preferentemente a mujeres de mediana edad (edad media: 35 años), tal como el caso que presentamos, siendo muy rara su manifestación en la infancia (6). Puede surgir en varias generaciones de una misma familia ya que se hereda de forma autosómica dominante con penetrancia incompleta (7). Se lo ha vinculado con diferentes alelos HLA, particularmente con una mayor prevalencia de HLA-DR3 y/o DR4 (8). Es condición indispensable la presencia de ISP para hacer su diagnóstico, pudiendo muchas veces ésta preceder a las otras endocrinopatías (enfermedad tiroidea autoinmune y/o DM tipo1), aun cuando su curso sea aparentemente indolente. Se considera que aproximadamente 75% de los casos de SPA tipo 2 se presentan con la combinación de falla adrenal y falla tiroidea (9). Puede asociar enfermedades autoinmunes menores como vitíligo, gastritis crónica atrófica, hipogonadismo hipergonadotrópico, etc. En orden decreciente, las enfermedades que forman parte del SPA tipo 2 son: ISP (100%), tiroiditis autoinmune (69%), DM (52%), vitíligo (11%), hipogonadismo (9%) (Tabla IV).
La Enfermedad de Addison se caracteriza por la producción insuficiente por parte de la glándula suprarrenal de cortisol, mineralocorticoides y andrógenos. Tiene una incidencia de 0,83 casos/100.000 habitantes y una prevalencia de 4 – 6 casos/100.000 habitantes, siendo su edad media de presentación los 40 años (10). Su etiología más prevalente en la actualidad es la adrenalitis autoinmune, con 70 a 90%. Otras causas mucho menos frecuentes son: infección por tuberculosis (principal causa del siglo pasado), hemorragia o infarto adrenal, otras infecciones (histoplasmosis, citomegalovirus, VIH, sífilis), enfermedad adrenal metastásica y secundaria a fármacos (5). Entre las principales manifestaciones clínicas de la insuficiencia adrenal se mencionan: astenia, hiperpigmentación, anorexia, hipotensión y trastornos gastrointestinales (Tabla V).

Suelen aparecer en forma insidiosa y es necesaria una destrucción de 90% de la corteza adrenal para que se manifiesten (5). Los síntomas pueden agruparse según el déficit que se produce:
*Déficit de glucocorticoides: hiperpigmentación de piel y mucosas, tal como el caso clínico que presentamos. Ésta es producida por el aumento del precursor del ACTH que produce ACTH y MSH (hormona estimulante de los melanocitos). La carencia de cortisol genera fatigabilidad muscular, hipoglicemia, síntomas digestivos como diarrea y/o estreñimiento, y síntomas psiquiátricos como irritabilidad e insomnio.
*Déficit de mineralocorticoides: hipotensión arterial, hipotensión ortostática, palpitaciones y síncope, manifestaciones presentes en el caso clínico que presentamos.

*Déficit de andrógenos: pérdida del vello axilar y pubiano, descenso de la líbido y oligomenorrea (11).
Aunque el diagnóstico de insuficiencia suprarrenal debe confirmarse con una valoración funcional del eje hipofisarioadrenal, en los casos de elevada sospecha y gravedad, debe iniciarse el tratamiento sin esperar los resultados de los exámenes diagnósticos (12). Para realizar el diagnóstico de enfermedad de Addison, además de las manifestaciones clínicas, es necesaria la determinación sérica de cortisol y ACTH.
Los valores normales de cortisol oscilan entre 5 y 20 ?g/dLen la mañana. Se acepta como criterio definitivo de insuficiencia adrenal un cortisol menor a 5 g/dL mientras que valores mayor o igual 18 g/dL excluyen el diagnóstico. La determinación asociada de ACTH no sólo permite apoyar el diagnóstico, sino que también ayuda a facilitar la distinción entre una insuficiencia adrenal primaria y secundaria. Se considera ISP a valores de ACTH elevados mayor a 100 pg/mL (el valor normal es de 10 a 100 pg/mL), mientras que en la insuficiencia suprarrenal secundaria los valores de ACTH son bajos o indetectables.Cuando se sospecha una insuficiencia suprarrenal y las determinaciones hormonales no definen claramente el trastorno, debe realizase una prueba de estimulación rápida con ACTH. En este examen diagnóstico se utilizan 250 ?g de cosintropina (1-24 ACTH) intravenoso y se mide la concentración de cortisol a los 30 minutos y a la hora. Se considera una prueba normal, en la que se generan niveles de cortisol mayor o igual a 18 ug/dL, excluyendo así el diagnóstico de ISP, mientras que una respuesta menor lo confirma (12,14).
Una vez confirmada la ISP debemos investigar su etiología. Para ello se solicita:
- Anticuerpos anti-suprarrenales: presentan elevada sensibilidad y especificidad teniendo carácter diagnóstico de adrenalitis autoinmune. Sus valores descienden con el tiempo, no siendo útiles para la monitorización del estado clínico y ni como guía terapéutica (4).
- Anticuerpos anti 21-hidroxilasa: presentes en 80% de las adrenalitis autoinmunes, no disponibles en nuestro medio (14).
- Estudios de imagen: Tomografía computada o resonancia magnética: las glándulas normales o atróficas bilateralmente son características de la adrenalitis autoinmune. En la etiología tuberculosa podemos encontrar calcificaciones, mientras que el agrandamiento glandular es característico de la enfermedad adrenal metastásica o neoplasias primitivas, hemorragias suprarrenales e infecciones micóticas y víricas (4).
- Otra paraclínica según la causa planteada: radiografía de tórax, PPD, VIH, VDRL, etc.
El tratamiento de la enfermedad de Addison consiste en reemplazar el glucocorticoide y el mineralocorticoide faltantes. La terapéutica de elección es la hidrocortisona, por su actividad glucocorticoidea, a dosis de 30 mg/día vía oral, 20 mg por la mañana y 10 mg por la tarde, con el objetivo de mantener el ritmo circadiano del cortisol; y la fludrocortisona, por su actividad mineralocorticoidea, a dosis de 0,05 a 0,2 mg por día (16). Deben administrarse las dosis mínimas necesarias, siendo la desaparición de los síntomas, la ausencia de hipotensión ortoestática y la normalización del ionograma los parámetros que nos indican que la dosificación del tratamiento es la correcta. Su efecto adverso más frecuente es el insomnio, el cual se previene administrando la última dosis de hidrocortisona en la tarde. Otras opciones de tratamiento son la prednisona o la dexametasona, pudiendo ser útiles en pacientes y situaciones concretas. La dosis habitual de prednisona es de 5 a 7,5 mg/día (13,15). Los pacientes deben ser educados para que lleven consigo un elemento que recuerde que son portadores de la enfermedad de Addison. En situaciones que aceleran el metabolismo hepático de la hidrocortisona como fiebre, infecciones, stress e interacciones medicamentosas (comitoina, barbitúricos, rifampicina), es necesario incrementar la dosis de hidrocortisona al doble y en algunos casos al triple, por tres días o más, según la evolución.

Pacientes con intolerancia digestiva alta, intervenciones quirúrgicas y/o traumas graves deben recibir hidrocortisona por vía intravenosa o intramuscular, y así evitar el desarrollo de una crisis adrenal aguda (5,13). A todos los pacientes con ISP de causa autoinmune se les debe determinar TSH, T4, anticuerpos antitiroideos y glicemia, ya que a un porcentaje no menor de éstos pacientes puede desarrollar una o más endocrinopatías adicionales. Entre 69 y 82% de los SPA tipo 2 presentan asociada tiroiditis autoinmune a forma de hipertiroidismo primario (Enfermedad de Graves) o hipotiroidismo primario (Tiroiditis de Hashimoto), siendo más prevalente la asociación enfermedad de Addison e hipotiroidismo (6). Generalmente, la enfermedad de Graves se desarrolla antes de la ISP, a una edad media de 31 años, mientras que la tiroiditis crónica se manifiesta simultáneamente o un año después del diagnóstico de enfermedad de Addison, con una media de edad de presentación de 39,6 años (6). Para realizar el diagnóstico de enfermedad tiroidea autoinmune es necesario dosificar los anticuerpos contra antígenos específicos tiroideos como antitiroglobulina, antiperoxidasa (antimicrosomales) y anti-receptor TSH, los cuales en ocasiones preceden por años a las manifestaciones clínicas de la enfermedad (16). La DM tipo 1 se produce entre 30 y 52% de los pacientes con SPA tipo 2. La edad media de presentación es menor a 28 años. Los pacientes con DM en el contexto del SPA tipo 2 tiene una alta frecuencia de anticuerpos anti-glutamato decarboxilasa, anti-islotes pancreáticos y antiinsulina positivos y puede presentarse de forma aguda o tener un inicio más insidioso como en la diabetes autoinmune latente del adulto (2,6). Es característico el desarrollo de otras enfermedades autoinmunes menores en el SPA tipo 2, se citan como ejemplo: hipogonadismo hipergonadotrópico, vitíligo, alopecia, hepatitis autoinmune, gastritis atrófica crónica y anemia perniciosa. Estas enfermedades se presentan con una frecuencia más baja en comparación con el SPAtipo 1 (6).
Por otro lado, el caso clínico que presentamos asocia insuficiencia ovárica prematura, que se define como el cese de la menstruación antes de los 40 años. Son criterios diagnósticos: amenorrea de más de cuatro meses en mujer menor de 40 años y concentraciones de FSH mayores a 40 mUL/mL y de estradiol menores de 20 ng/mL (hipogonadismo hipergonadotrópico), con una diferencia entre 30 y 45 días. Presenta distintas causas, siendo la etiología autoinmune la responsable de 15 a 30% de las fallas ováricas prematuras (17). Son característica de esta entidad; la presencia de autoanticuerpos anti ovarios, infiltración linfocítica ovárica y la asociación con otras enfermedades autoinmunes endocrinológicas (18). Forma parte del SPAtipo 1 en aproximadamente el 60% de los casos, mientras que su presentación en el tipo 2 es mucho menor, pudiendo alcanzar un 10% (6).
La detección temprana de la enfermedad endocrinológica y su tratamiento antes de que produzca una morbimortalidad significativa son claves para un manejo exitoso del paciente con un SPA. El tratamiento de las distintas endocrinopatías asociadas no varía del que se haría ante cada afección en forma asilada. Sin embargo, vale la pena recordar que en los pacientes con hipotiroidismo y enfermedad de Addison, se debe primero normalizar la función adrenal y luego la tiroidea, ya que de lo contrario puede producirse una crisis adrenal aguda. Por otra parte, una reducción en los requerimientos de insulina puede ser el primer signo de la enfermedad de Addison en pacientes con DM tipo 1. Por lo tanto, antes de iniciar el tratamiento con tiroxina o simplemente modificar la dosis de insulina, es prudente investigar la posible coexistencia de una insuficiencia suprarrenal subyacente. Está empezando a plantearse la posibilidad de iniciar tratamientos profilácticos antes de que aparezcan las diferentes enfermedades del SPAy es un objetivo no muy lejano instaurar tratamientos que permitan prevenir o al menos frenar la progresión de estas enfermedades (1,3,6).
Conceptos clave
En todo paciente que se realiza el diagnóstico de ISP de causa autoinmune, debemos buscar la presencia de otras endocrinopatías inmunomediadas dada su frecuente asociación.
Existen 4 tipos de SPA, siendo el tipo 2 el más frecuente de los descritos.

La detección temprana de auto-anticuerpos órgano específicos en pacientes con una endocrinopatía autoinmune, facilita la identificación de pacientes con riesgo de desarrollo futuro de otras endocrinopatías autoinmunes.
El descubrimiento precoz de una endocrinopatía asociada y su tratamiento antes de que se haga sintomática, son claves en el manejo exitoso del paciente con un SPA, mejorando así su pronóstico funcional y vital.
Actualmente, el abordaje de estas enfermedades se limita a la terapia de reemplazo farmacológico. Sin embargo, el progreso en el conocimiento de los mecanismos inmunológicos en juego, generan como meta a futuro establecer tratamientos que permitan prevenir, o al menos frenar, la progresión a un daño irreversible en múltiple órganos endócrinos.
Referencias Bibliográficas
1. Neufeld M, Maclaren NK, Blizzard RM. Two types of autoimmune Addison’s disease associated with different polyglandular autoimmune (PGA) syndromes. Medicine 1981; 60: 355-362.
2. Betterle C et al. Autoimmune adrenal insufficiency and autoimmune polyendocr ine syndromes : autoantibodies, autoantigens, and their applicabilbility in diagnosis and disease prediction. Endocr Rev 2002; 23: 327-364.

3. Molina Garrido MJ. Síndrome pluriglandular autoinmune. Revisión. An. Med. Interna (Madrid) 2007; 24(9): 445-452.
4. Fernández Serrano F, et al. Insuficiencia suprarrenal primaria como primera manifestación de un síndrome pluriglandular autoinmune, SEMERGEN 2005;31(1):31- 34.
5. Lovas K, Husebye E. Addison s disease. Lancet. Junio 2005(9476);365:2081-2061.
6. Betterle C, Lazzarotto F, Presotto F. Autoimmune polyglandular syndrome Type 2: the tip of an iceberg? Clin Exp Immunol 2004;137:225–233.
7. Eisenbarth GS, Wilson PN, Ward F, Buckley C, Lebovitz H. The polyglandular failure syndrome: disease inheritance, HLA-type, and immune function studies in patients and families. Ann Int Med 1979; 91:528-533.
8. Badenhoop K,Walfish PG, Rau H, Fisher S Nicolay A, Bogner U, Schleusener H, Usadel KH. Susceptibility and resistance alleles of human leukocyte antigen (HLA) DQA1 and DQB1 are shared in endocrine autoimmune disease. J Clin Endocrinol Metab 1995; 80: 2112-2117.
9. Graves L, Klein R, Walling A. Addisonian crisis precipitated by thyroxine therapy: a complication of type 2 autoimmune polyglandular syndrome. Southern Medical Journal. Agosto 2003;96(8):824-827.
10. Oelkers W. Adrenal insufficiency. Current-concepts. N Engl J Med 996;335:1206-12.
11. Werbel SS, Ober KP. Acute Adrenal insufficiency. Endocrinol Metab Clin North Am 1993; 22:303-328.
12. O. González-Albarrán, R. García Robles. Insuficiencia suprarrenal primaria. Medicine. 2000; 8(22): 1141-1147.
13. J.J. Corrales Hernández. Insuficiencia suprarrenal crónica primaria: enfermedad de Addison. Insuficiencia suprarrenal aguda. Hipoaldosteronismos hiporreninémicos e hiperreninémicos. Pseudohipoaldosteronismos. Medicine. 2008;10(15):986-996.
14. O. González-Albarrán, R. García Robles. Protocolo se sospecha de insuficiencia suprarrenal. Medicine. 2000; 8(22): 1168-1169.
15. Ten S, New M, Maclaren N. Addison´s disease 2001. J Clin Endocrinol Metab. Julio 2001;86 (7):2909-2922.
16. G. Martínez Díaz-Guerra, A. Serraclara Pla, E. Jódar Gimeno y F. Hawkins Carranza. Patología tiroidea. Clasificación. Evaluación de la función tiroidea. Anticuerpos antitiroideos. Tiroglobulina. Imagen en tiroides: ultrasonografía, gammagrafía, TAC y PET. Punción-aspiración de tiroides. Medicine. 2008; 10(14):889-887.
17. Scaglia, J. Premature Ovarian Failure. Rev Argent Endocrinol Metab. 2007: 44:242-247.
18. Hoek A, Schoemaker, Drexhage A. Premature Ovarian Failure and Ovarian Autoimmunity. Enndocrine Reviews.1997; Vol. 18. No 1:107-132.


Más notas de la edición 14
Lee nuestra última edición publicada en Marzo 2026, Edición número 175

Notas relacionadas a Enfermedad de Addison como presentación de un...

