



Diabetes mellitus por mutación en el gen de glucokinasa. Caso clínico
Felipe Pollak C.1, Marcela Lagos L.2, José L. Santos M.1, Helena Poggi2, Abraham Urzúa C.2, Hana Rumié C.3
1Departamento de Nutrición, Diabetes y Metabolismo, Facultad de Medicina, Pontificia Universidad Católica de Chile. Santiago, Chile
2Laboratorio Biología Molecular, Facultad de Medicina, Pontificia Universidad Católica de Chile. Santiago, Chile
3Unidad de Endocrinología Pediátrica, Complejo Asistencial Dr. Sótero del Río. Santiago, Chile
Correspondencia: Felipe Pollak C. fpollak@med.puc.cl Bernarda Morín 488, Providencia, Casilla 168, Correo 55 Santiago – Chile Tel.: (56-2) 2753 5520 revmedchile@smschile.cl
Revista médica de Chile versión impresa ISSN 0034-9887
Rev. méd. Chile vol.145 no.9 Santiago set. 2017
http://dx.doi.org/10.4067/s0034-98872017000901203
Abstract
Maturity-Onset Diabetes of the Young (MODY) refers to a heterogeneous group of monogenic diabetes. Unlike other types of MODY characterized by genetic defects in transcription factors, MODY 2 is triggered by metabolic alterations caused by mutations of glucokinase (GCK), the first enzyme of the glycolytic pathway. We report a three-generation Chilean family with multiple cases affected with this disease. The index case is a patient who presented severe neonatal hyperglycemia (831 mg/dl, without ketosis) requiring continuous infusion of insulin, which was suspended after 48 hours with normalization of blood glucose. Subsequently, continuous glucose monitoring at 4 months of age revealed 47% of tissue glucose levels above 140 mg/dl, with fasting glucose levels between 120 and 166 mg/dl. The genetic analysis revealed a previously reported mutation in heterozygous state of the GCK gene (c.148C>T; p.His50Tyr). This mutation was also identified in more than one affected relative in the last two generations, with a transmission pattern suggestive of dominant inheritance. GCK gene sequencing led to a correct molecular diagnosis of MODY 2 while bioinformatic analysis indicated the possible molecular causes of the enzyme dysfunction. The knowledge of the molecular diagnosis allowed an adequate medical treatment for this disease.
Key words: Maturity-Onset Diabetes of the Young; Type 2; glucokinase; mutation; autosomal dominant
Introducción
Entre las Diabetes Mellitus (DM) producidas por defectos genéticos de la célula beta pancreática, las más mencionadas corresponden al grupo de MODY (Maturity-Onset Diabetes of the Young)1. Se caracterizan por mutaciones de transmisión autosómica dominante que alteran la secreción insulínica, con hiperglicemia de grado variable, generalmente de aparición precoz (antes de los 25 años). No presenta asociación con Síndrome Metabólico o anticuerpos específicos. Se calcula que este grupo podría corresponder a un 5% del total de pacientes diabéticos.
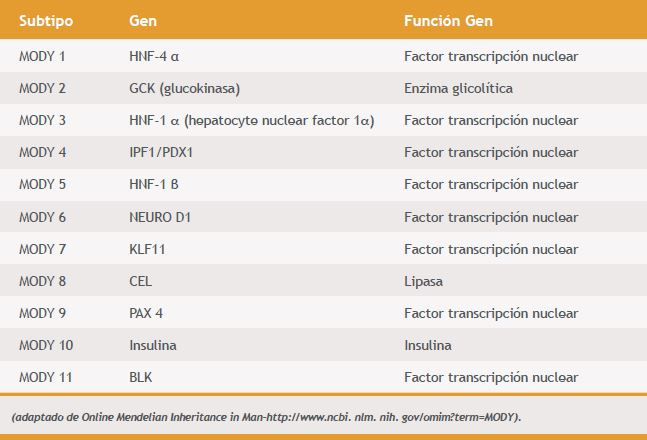
Se han descrito a lo menos 11 genes diferentes para la enfermedad (Tabla 1). Los más frecuentes son el estado heterocigoto para el gen de la Glucokinasa (GCK) o MODY tipo 2 y para el gen de HNF-1 α (hepatocyte nuclear factor 1-α) o MODY tipo 32.
Tabla 1. Tipo de MODY y genes asociados
La GCK pertenece a la familia de las hexokinasas, cuya función es fosforilar a hexosas (D-glucosa, D-manosa y D-fructosa). Cataliza la fosforilación de glucosa a glucosa-6-fosfato (G-6-P) en la primera reacción de la glicólisis, tanto en la célula beta como en hepatocitos, siendo clave en el metabolismo de los hidratos de carbono y de la homeostasis de la glucosa. Ejerce un rol fundamental en el control de la secreción de insulina, ya que ésta depende de los niveles intracelulares de ATP generados a partir de G-6-P.
Hasta el momento se han descrito más de 700 mutaciones para el gen de la GCK, presentando mayor prevalencia en países como Italia, Francia, Reino Unido y España3.
La pérdida de función en el gen de GCK produce una disminución de la fosforilación, y secundariamente de la sensibilidad de la célula beta a la glucosa, lo que produce elevación del umbral glicémico que estimula la secreción de la insulina. Además disminuye la síntesis hepática de glucógeno. Esto se traduce en un aumento discreto de la glicemia a niveles de 100 a 145 mg/dl y de aproximadamente 50 mg/dl después de una sobrecarga a la glucosa. En casos de estado homocigoto puede presentarse como Diabetes Neonatal con requerimientos de insulina4.
Clínicamente se caracteriza por niveles de hemoglobina glicosilada A1c (HbA1c) que rara vez sobrepasan a 7,5% y sin necesidad de fármacos hipoglicemiantes. El riesgo de complicaciones microvasculares es bajo5. La presencia de obesidad y factores de riesgo cardiovascular asociados es infrecuente.
Su comienzo en la vida es precoz, iniciándose ya en la vida intrauterina, donde los menores niveles de insulinemia generan con frecuencia bajo peso de nacimiento. Aún así, la falta de sintomatología puede derivar en un diagnóstico a mayor edad, en la juventud o adultez.
Se describe que mutaciones en este gen, se encuentran en el 50% de los casos de MODY. Paralelamente puede explicar 2-5% de los casos de DM Gestacional, debiendo sospecharse frente a hiperglicemia leve en el primer trimestre del embarazo, a temprana edad, y en ausencia de obesidad y elementos de resistencia a la insulina6.
Finalmente existe heterogeneidad en su presentación, describiéndose que los portadores de la mutación G261R (exón 7) tienen mayores niveles de glicemia, incluso presentando hiperglicemia neonatal7.
Caso Clínico
Paciente sexo masculino, con historia de embarazo fisiológico, parto eutócico, peso nacimiento 3.110 g (-0,27 ds), longitud 52 cm (-1,59 ds) y apgar 3-5-6. Cursa con asfixia neonatal e hipertensión pulmonar severa manejada con hipotermia, ventilación de alta frecuencia y óxido nítrico. A las 72 h de vida presenta hiperglicemia hasta 831 mg/dl, sin cetosis, que requirió infusión de insulina hasta un máximo de 0,5 U/kg/h. A las 48 h se suspende la insulina endovenosa con rápida normalización de glicemia. No presentó nuevos episodios de hiperglicemia. Dado de alta en buenas condiciones a los 15 días de vida.
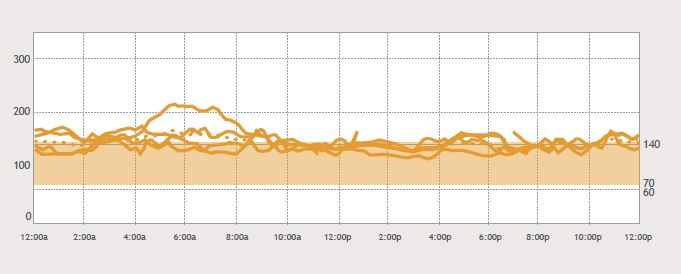
A los 4 meses de vida se realiza monitoreo continuo de glucosa (5 días de duración) con un 47% de glicemias tisulares mayores a 140 mg/dl. y glicemias de ayuno de 120 a 166 mg/dl (Figura 1).
Figura 1. Monitoreo continuo de glucosa, caso índice.

Su madre no presentaba antecedentes de importancia. El padre presentaba glicemia de ayuno en 138 mg/dl, insulinemia 7,7 uUI/ml y HbA1c de 6,1%.
Su hermano de 11 años presentaba glicemia ayuno de 109 mg/dl, insulinemia basal 7,2 uUI/ml y HbA1c de 7%.
Su tía paterna tenía como antecedente DM gestacional en sus 2 embarazos, a los 19 y 27 años respectivamente. En ambas oportunidades requirió uso de insulina para control adecuado. Sus hijos tuvieron peso de nacimiento normal. El mayor de ellos presentaba glicemia alterada en ayuno.
Su abuela y bisabuela paterna son diabéticas, esta última insulinorequirente, con hipertensión arterial y retinopatía diabética no proliferativa moderada como complicación.
Todos los familiares evaluados presentaban Índice de Masa Corporal normal.

Se sospecha DM monogénica por defecto de secreción de la célula beta y se maneja con medidas dietéticas y controles periódicos de antropometría y HbA1c.
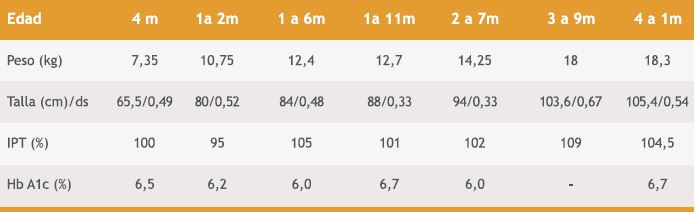
Su seguimiento se detalla en la Tabla 2.
Tabla 2. Seguimiento caso índice
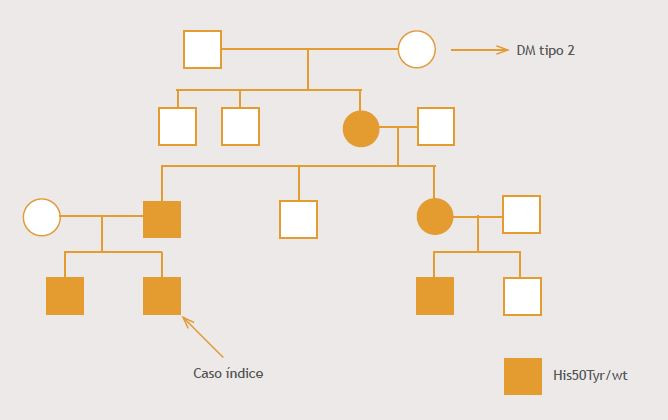
Se realiza el estudio molecular del gen GCK, que identificó la variante c.148C>T, p.His50Tyr (a excepción de bisabuela), como lo muestra el genograma (Figura 2).
Figura 2. Genograma caso clínico.

Metodología
Para implementar la técnica de diagnóstico genético de DM por mutación en el gen de GCK, se invitó a especialistas en Diabetología y de Endocrinología pediátrica de diferentes centros a referir pacientes con sospecha clínica para estudio genético. De estos casos se seleccionó uno de ellos para presentación. El estudio fue aprobado por el Comité de Ética de la Facultad de Medicina de la Pontificia Universidad Católica de Chile.
El estudio genético fue realizado en el Laboratorio de Biología Molecular del Departamento de Laboratorios Clínicos de la Facultad de Medicina de la Pontificia Universidad Católica de Chile. Previa firma de consentimiento informado, se obtuvo muestras de sangre de los pacientes, y se procedió a la extracción de DNA genómico, por medio de QIAmp DNA Blood Mini Kit.
En la Reacción en Cadena de la Polimerasa (PCR), los primers utilizados para la amplificación de cada exón se obtuvieron de la literatura científica8.
Finalmente se realizó secuenciación bidireccional de tipo Sanger utilizando un equipo de electroforesis capilar ABI PRISM® 3130 (Applied Biosystems).
Los electroferogramas obtenidos fueron comparados con las secuencias de referencia del gen GCK (NG_008847.1 y NM_000162.3). Las mutaciones fueron reportadas de acuerdo a la nomenclatura del Human Genome Variation Society (hgvs.org)9.

La predicción bioinformática mediante los programas Poliphen-2, SIFT y modelos de predicción in silico indicaron que la variante encontrada afectaría la función enzimática a través de la pérdida de puentes de hidrógeno e interacciones iónicas entre aminoácidos.
Discusión
Se presenta un caso indíce atípico con hiperglicemia significativa asociada a estrés en período neonatal. Esta situación revierte a los pocos días, presentando estabilización de sus glicemias aunque persistiendo con glicemias alteradas a lo largo de su crecimiento.
La variante confirmada para esta familia es c.148C>T, p.His50Tyr. Tiene carácter patogénico, dado que produce una disminución de la actividad enzimática de GCK y ha sido reportada en la literatura10,11.
Si bien la marcada hiperglicemia neonatal no es característica de la enfermedad, como explicación es posible mencionar el estrés inducido en relación a la asfixia neonatal, en un paciente con defecto de la secreción y dificultad para compensar adecuadamente la insulinoresistencia transitoria. Por otro lado, y tal como se mencionó previamente, se ha reportado que algunas variantes pueden presentarse de forma particular, lo que también puede colaborar a explicar la magnitud de la glicemia inicial. La evolución posterior al período neonatal y la presentación en los familiares afectados es la descrita para esta patología.
Su bisabuela, con estudio negativo para mutaciones en GCK, presenta clínica concordante con una DM tipo 2, patología prevalente en nuestro medio.

Aún tratándose de un cuadro de características “no agresivas”, el estudio genético se justifica para el diagnóstico diferencial, manejo clínico adecuado, pronóstico y consejo genético de los afectados (aproximadamente 50% de los hijos heredan la mutación). En la familia estudiada, alguno de los afectados recibían fármacos hipoglicemiantes, los que considerando la precisión diagnóstica fueron suspendidos, manteniendo niveles estables de glicemia y HbA1c.
Finalmente, recomendamos realizar estudio genético a pacientes con hiperglicemia leve o HbA1c levemente elevada (< 7,5%), persistente en el tiempo, ausencia de complicaciones específicas, estudio de autoinmunidad negativo y presencia de DM a lo menos en 2 generaciones.
AGRADECIMIENTOS: A Fondecyt 1150416
Referencias Bibliográficas
1. American Diabetes Association. Diagnosis and Classification of Diabetes Mellitus. Position Statement. Diabetes Care 2017; 40 (Suppl. 1): S11-S24.
2. Fajans S, Bell G. MODY: History, genetics, pathophysiology, and clinical decision making. Diabetes Care 2011; 34: 1878-84.
3. Human Gene Mutation Database, http://www.hgmd.org/
4. Codner E, Deng L, Pérez-Bravo F, Román R, Lanzano P, Cassorla F, et al. Glucokinase mutations in young children with hyperglicemia. Diabetes Metab Res Rev 2006; 22: 348-55.
5. Steele AM, Shields BM, Wensley KJ, Colclough K, Ellard S, Hattersley AT. Prevalence of vascular complications among patients with glucokinase mutations and prolonged, mild hyperglycemia. JAMA 2014; 311 (3): 279-86.
6. Watanabe RM, Black MH, Xiang AH, Allayee H, Lawrence JM, Buchanan TA. Genetic of Gestational Diabetes Mellitus and Type 2 Diabetes. Diabetes Care 2007; 30 (suppl. 2): S134-40.
7. Cuesta-Muñoz AL, Tuomi T, Cobo-Vuilleumier N, Koskela H, Odili S, Stride A, et al. Clinical Hetrogeneity in Monogenic Diabetes Caused by Mutations in the Glucokinase Gene. Diabetes Care 2010; 33: 290-2.
8. Yokota I, Moritani M, Nishisho K, Miyoshi T, Kotani Y, Kagami S. Detection of glucokinase gene defects in non-obese Japanese children diagnosed with diabetes by school medical examinations. Endocr J 2011; 58 (9): 741-6.
9. http://www.hgvs.org/
10. Massa O, Meschi F, Cuesta-Muñoz A, Caumo A, Cerutti F, Toni S, et al; Diabetes Study Group of the Italian Society of Paediatic Endocrinology and Diabetes (SIEDP). High prevalence of glucokinase mutations in Italian children with MODY. Influence on glucose tolerance, first-phase insulin response, insulin sensitivity and BMI. Diabetologia 2001; 44 (7): 898-905.
11. Mantovani V, Salardi S, Cerreta V, Bastia D, Cenci M, Ragni L, et al. Identification of eight novel glucokinase mutations in Italian children with maturity-onset diabetes of the young. Hum Mutat 2003; 22 (4): 338.
Los autores no declaran conflicto de intereses.
Más notas de la edición 79

Lee desde Issuu nuestra última edición publicada en Julio 2024, Edición número 155

Notas relacionadas a Diabetes mellitus por mutación en el gen de...

