

Déficit de biotinidasa o Déficit múltiple de carboxilasas
Dra. Ligia M Marcos Plasencia; Dra. Laritza Martínez Rey; Dra Ana Jenny Pérez Torres
- Esp de I G en Pediatría y de IIG en Nutrición. Dpto. de Clínica de la Nutrición del INHA
** Esp en Genética Clínica. CNGM
***Esp en MGI, Polic. Giron, Ciudad Habana Ministerio de Salud Pública, República de Cuba Trabajo de revisión bibliográfica.
Resumen
Recientemente se ha dado inicio a la pesquisa neonatal para Déficit de Biotinidasa o déficit de carboxilasas múltiples en el país. Para el tratamiento y seguimiento de estos casos se hace necesario que los equipos multidisciplinarios provinciales responsables estén preparados para ello. Con este material se trata de contribuir a la capacitación así como mediante cursos impartidos en las diferentes provincias. Esta enfermedad es susceptible de tratamiento médico con lo cual se puede evitar graves daños al subsistema nervioso o incluso la muerte. El diagnóstico y tratamiento deben ser precoces para lograr el objetivo.
Historia de la Biotina
- Fue descubierta por Gyorgy entre 1931 y 1940, como factor protector contra la posible toxicidad para el humano de la clara de huevo cruda.
- En 1936 fue aislada en forma cristalina de la yema de huevo por Kogl y Tonnis.
- En 1942, Du Vigneaud y cols. publicaron su estructura química.
- En 1943, Harris la sintetizó.
Información previa
Características de la biotina:
- Vitamina esencial hidrosoluble necesaria para el mantenimiento de la actividad metabólica normal.
- Perteneciente a las vitaminas del complejo “B”, conocida anteriormente como vitamina “H” o vitamina 8.
- Contiene azufre, es un derivado imidazólico de amplia distribución en la naturaleza.
- Es sintetizada por la flora intestinal, pero su mecanismo de absorción es aún discutido.
- Una vez absorbida es transportada por la sangre unida a la albúmina y a las globulinas.
- Constituye parte integral (grupo prostético) de algunas enzimas transportadoras de grupos carboxilos (carboxilasas), formando una amida con el grupo amino (NH2) de un residuo del aminoácido lisina de la enzima carboxilasa, nombrada biotinil-lisina.
- Las enzimas carboxilasas son 4 y las reacciones que catalizan son las siguientes:
a) Paso inicial en la biosíntesis citoplasmática de los ácidos grasos:
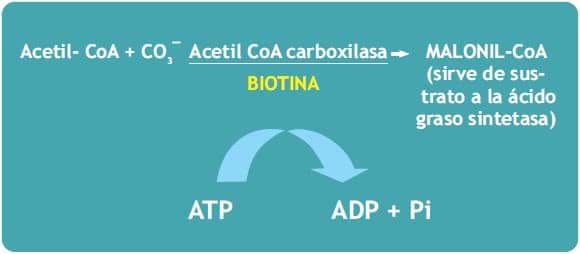
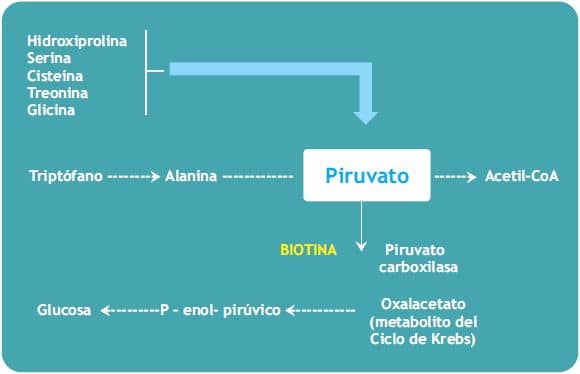
b) La glucogénesis (biosíntesis de glucosa a partir de compuestos no glucídicos). La Piruvato carboxilasa cataliza la incorporación de bicarbonato al piruvato para formar oxalacetato, un intermediario del ciclo de Krebs. En los tejidos con capacidad gluconeogénica (hígado y riñón) el oxalacetato puede ser convertido en glucosa.
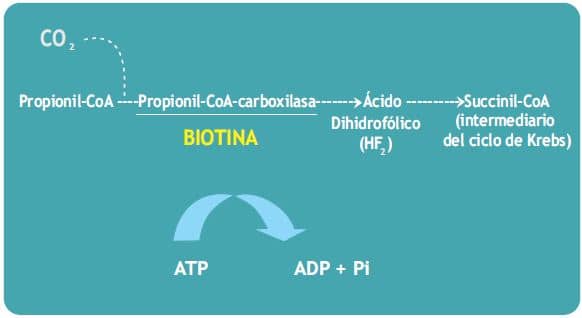
c) La formación de Succinil-CoA y ácido oxalacético, metabolitos del ciclo de Krebs. La Propionil-CoA-carboxilasa cataliza la incorporación de bicarbonato a la propionil-CoA para formar Succinil-CoA, que penetra en el ciclo de Krebs.
d) El catabolismo del aminoácido leucina y de ciertos compuestos isoprenoides. La metilcrotonil-CoA-carboxilasa, cataliza un paso esencial en la degradación del aminoácido ramificado Leucina.

8. Otras carboxilasas dependientes de biotina:
- Geramil-CoA carboxilasa de Biotina
- Aminocarboxilasa
9. Es cofactor de otras enzimas no carboxilasa:
- Metil-malonil-CoA-descarboxilasa
- Oxaloacetato descarboxilasa
- Transcarboxilasas dependientes de Biotina (participan en la fermentación bacteriana)
10. Otras funciones:
- Crecimiento celular
- Homeostasis de la glucosa (en dosis farmacológicas, reduce la glicemia de ayuno del diabético insulino – dependiente cuando no se le administra insulina).
- Síntesis de DNA
11. Causas del déficit de biotina:
a. Congénita
- por déficit de holocarboxilasas sintetasa
- por déficit de biotinidasa
b. Adquirida
- por esterilización intestinal
- por ingestión prolongada de huevos crudos, pues la clara contiene avidina, glucoproteína termolábil que se une a la biotina e impide suabsorción
- en la nutrición parenteral sin suplemento de biotina en pacientes con síndrome de intestino corto u otras causas de malabsorción
- en individuos con tratamiento anticonvulsivante a largo plazo, que disminuye los nivelesplasmáticos de la biotina, aunque no está claro el mecanismo de la depleción. Los medicamentos involucrados son: fenobarbital, difenilhidantoína,carbamacepina, primidona (todas con un fragmento carbamida NH-CO- en su estructura, igual que la biotina).
12. Estructura de la biotina:
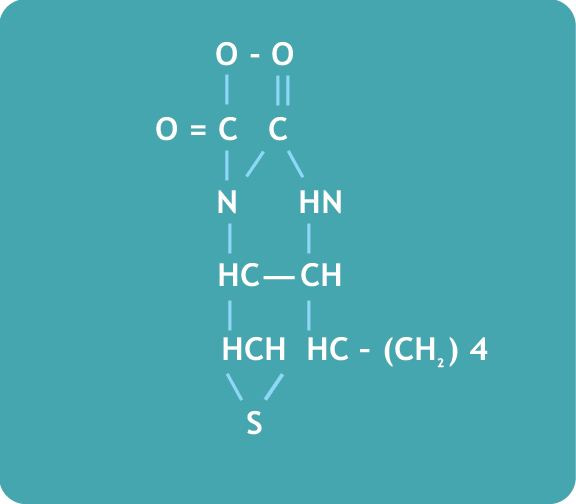
13. Síntomas en caso de deficiencia adquirida:
Aparecen en forma gradual con adelgazamiento, cambio en la coloración del cabello, dermatitis no pruriginosa, mialgias, somnolencia, depresión e hiperestesia. Más tarde anorexia, náuseas, anemia, hipercolesterolemia. Todo desaparece de 2 a 5 días de iniciar el tratamiento sustitutivo.
14. Niveles de ingestión seguros y adecuados para el metabolismo normal, propuestos por la RDA,10 edición , 1989:
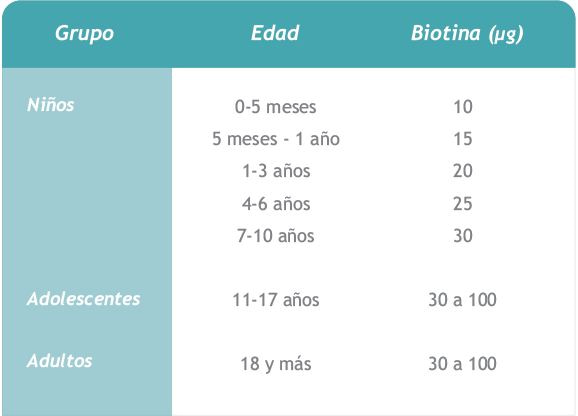
Los casos de deficiencia de esta vitamina son extremadamente raros, ya que la dieta habitual suele cubrir con amplio margen de seguridad los niveles de ingestión propuestos.
15. Usos:
- Para el déficit de biotinidasa o de carboxilasas
- Otros usos: para estados carenciales de biotina como acné, eczema seborreico, fragilidad ungueal, eritrodermia, dermatitis exfoliativa
16. Modo de empleo: oral (cdtas o cap) o IM (amp).
Definición de la enfermedad Congénita
Trastorno metabólico, perteneciente al grupo de las acidurias orgánicas, de herencia autosómica reseciva, poco frecuente, cuya alteración fisiopatológica radica en un déficit en la actividad de la enzima biotinidasa que debe separar la biotina de la biocitina, con el consecuente déficit de carboxilasas, o también por un déficit de la enzima holocarboxilasa sintetasa. La ausencia de la biotinidasa resulta en la ausencia de biotina. La falta de las carboxilasas mitocondriales lleva al acúmulo de algunos metabolitos y a la ausencia de otros, lo que produce eventos epilépticos.

Los neonatos con eventos epilépticos debidos a la falta de biotinidasa usualmente tienen alteraciones cutáneas, alopecía parcial o total, y conjuntivitis persistente.
Las carboxilasas juegan su papel en la gluconeo génesis, en la síntesis de ácidos grasos y en el catabolismo de los aminoácidos.
Su incidencia:
Globalmente oscila entre 1 por 27 000 y 1 por 277 000 nacidos vivos.
Aunque su incidencia es baja es un proceso de alta morbimortalidad, para el que existe un tratamiento efectivo.
Fisiopatología de enfermedad congénita
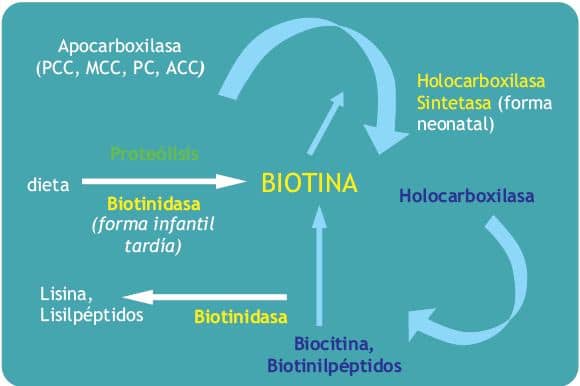
La biotina actúa como una coenzima en el metabolismo de las carboxilasas, que son:

- Propionil-CoA-carboxilasa (PCC)
- Beta-metil-crotonil-CoA-carboxilasa (MCC)
- Piruvato-carboxilasa (PC)
- Acetil CoA-carboxilasa (ACC)
Cuadro Clínico
Es amplio e inespecífico, lo que dificulta el diagnóstico clínico, que se hace generalmente tardío o no se hace. Con signos y síntomas neurológicos y dermatológicos. Suele acompañarse de acidosis láctica con cetosis.
En el período neonatal: Debida al déficit de la enzima holocarboxilasa sintetasa, algunos cuadros pueden aparecer en este período con estado de acidosis metabólica grave que puede llevar a la muerte con convulsiones, hipotonía muscular, alopecía, dermatitis, orina con olor a orina de gato, inmunodeficiencia, atrofia óptica. Con tratamiento revierte por completo. Aquí hay ausencia total de las holocarboxilasas. Se le adjudica responsabilidad en el síndrome de muerte súbita infantil.
En períodos posteriores (forma tardía): Debida al déficit de la enzima biotinidasa, con deterioro progresivo, ataxia, convulsiones, retardo del desarrollo psicomotor, déficit auditivo, dermatitis, alopecía (cabello y vello corporal), acidosis metabólica, atrofia óptica e inmunodeficiencia.
Muerte súbita. El daño neurológico es irreversible. Aquí disminuyen progresivamente las holocarboxilasas por la ausencia total de biotina, pero en los primeros tiempos el niño no ha repletado las reservas de biotina del nacimiento.
Diagnóstico
En orina:
aumento de la excreción de,

- ácido 3 hidroxi isovalérico,
- ácido metilcítrico,
- ácido 3 hidroxipropiónico y
- ácido 3 metil crotonil glicina
- cuerpos cetónicos en orina (lactato)
En sangre:
- Equilibrio ácido básico por gasometría: acidosis
- Determinación de cuerpos cetónicos en sangre: cetosis
- presencia de ácido láctico
- Aa: aumento de la alanina
- Disminución de la carnitina
Específico:
- medición de la actividad de la biotinidasa plasmática (puede ser indetectable o muy baja)
- actividad de las carboxilasas en leucocitos (linfocitos) y fibroblastos en sangre periférica (puede haber o no, disminución de la actividad de alguna de ellas o de todas)
Screening neonatal:
Por el UMTEST Biotinidasa, en muestra de sangre seca en papel de filtro recogida del talón del neonato a los 5 días de vida, que demuestra la actividad de la biotinidasa sérica. Se utiliza la misma muestra recogida para UMTEST PKU.
Prenatal:
No se realiza en Cuba.
Valores Normales
En sangre
| Lactato sérico | 0,67 a 1,80mM |
En orina
| Lactato Urinario | |
| 3-OH-Propiónico | |
| 3-OH-isovalérico | de 0 a 46 mMol/Mol creat. |
| Metil crotonil glicina | 0 |
| Meil citrato | 0 a a 12mMol/Mol creat. |
.
Confirmación
Se repite la prueba de UMTEST Biotinidasa una vez ingresado el paciente.
Se toma muestra de sangre y orina desde que el niño llega al hospital para:

- * determinar ácidos orgánicos en orina
- * determinar actividad de biotinidasa plasmática
- * determinar carboxilasas linfocitarias
Tratamiento
Debe ser precoz, es sustitutivo e impide la expresión clínica de la enfermedad, pues aún no se han depletado las reservas de biotina con que nace el neonato y no se acumulan los metabolitos intermediarios.
En edades posteriores es parcialmente efectivo, pues no revierte el daño neurológico ya instaurado. No obstante está indicado comenzar tratamiento en cualquier época de la vida en que se haga el diagnóstico, pues se logra una mejoría notable de muchos de los síntomas y se impide que continúe el deterioro del SN.
La acción de la biotina es rápida y produce una mejoría notable del estado general, se controlan las crisis convulsivas (clínicas y eléctricas) y se resuelven las lesiones cutáneas.
La biotina no tiene interacciones con otros medicamentos.
Los requerimientos de energía, proteínas, macronutrientes y micronutrientes se prescriben según sexo, edad y peso. No es necesario restricción proteica.

Al inicio: biotina: 20 mg/kg/día. En los casos no favorables la dosis puede llegar a los 40 mg/kg/día.
Posteriormente: * Biotina : 10 mg/kg/día.
Compuestos existentes en el mercado
Alimentos ricos en biotina: hígado de res, riñón, pollo, pescado, huevo entero, guisantes (chícharos, garbanzos, lentejas), maní, chocolate, cereales integrales, vegetales de color verde intenso, frutas cítricas y levaduras. Es bastante estable, el 70% o más de ella permanece después de su cocción, no se descompone por tratamiento ácido ni básico, pero se inactiva por el peróxido de hidrógeno y por aceites y grasas rancios.
Referencias Bibliográficas
1. ColomboCM, CornejoEV, RaimannEB: Errores innatos en el metabolismo del niño. Comité de publicaciones científicas. Vicerectoria de asuntos académicos. Univercidad de Chile.1999.
2. Couce PicoML, Martinon TorresF, CastiñeirasDE, Alonso Fdez JR, FragaJM: Deficiencia de biotinidasa: Importancia de su diagnóstico neonatal y tratamiento precoz. An Esp. Pediatric. 1999; 50: 504-506.
3. Zschocker/Hoffmann: Vademecum Metabolicum. Manual of Metabolic Pediatrics. Milupa. 1 ed. Stuttgart: Schattauer, 1999.
4. OMIM ENTRY 253260. Multiple carboxylase deficiency.
5. Campistol J, VilasecaMA, Ribes A, Riudor E. Deficit de biotinidasa. Forma de presentación y respuesta al tratamiento. An Esp. Pediatric 1996; 44: 389-392.
6. Porrata C y col. Recomendaciones nutricionales y guías de alimetación para la población cubana. Instituto de Nutrición e Higiene de los Alimentos. Editorial Pueblo y Educación.1996
7. Cáceres A, Hdez M, Muñoz J, Rguez A: Las vitaminas en la nutrición humana. Ayuntamiento San Bartolomé de Tirajana. Las Palmas de Gran Canaria. 1999. P 44-48.

Más notas de la edición 15
Lee desde Issuu nuestra última edición publicada en Septiembre 2025, Edición número 169

Notas relacionadas a Déficit de biotinidasa o Déficit múltiple de...

