
Daño hepatocelular, proteinuria y autoinmunidad: ¿enfermedad multisistémica o coincidencia de enfermedades?
CASO CLÍNICO
Patricio Herrera1 – Alex Ruiz2 – Daniel Carpio3 – Leopoldo Ardiles1
1 Unidad de Nefrología, Instituto de Medicina, Universidad Austral de Chile. Valdivia, Chile
2 Unidad de Gastroenterología, Instituto de Medicina, Universidad Austral de Chile. Valdivia, Chile
3 Instituto de Anatomía, Histología y Patología, Universidad Austral de Chile. Valdivia, Chile
Revista médica de Chile versión impresa ISSN 0034-9887 Rev. méd. Chile vol.146 no.6 Santiago jun. 2018 http://dx.doi.org/10.4067/s0034-98872018000600808
Correspondencia a: Leopoldo G. Ardiles, Laboratorio de Nefrología, Facultad de Medicina, Universidad Austral de Chile, Valdivia, Chile. leopoldoardiles@gmail.com
Los autores declaran no tener conflictos de interés.
Bernarda Morín 488, Providencia, Casilla 168, Correo 55
Santiago – Chile
Tel.: (56-2) 2753 5520
revmedchile@smschile.cl
Abstract
We report a 19 years old male presenting with knee pain, elevated liver enzymes and proteinuria. Further investigation found positive antinuclear and anti-smooth muscle antibodies and a liver biopsy revealed the presence of an autoimmune hepatitis. Treatment with corticosteroids and azathioprine was started, resulting in normalization of liver enzymes but proteinuria persisted and a kidney biopsy disclosed a focal segmental glomerulosclerosis. The use of lisinopril resulted in a significative reduction of proteinuria and, after 30 months of follow up, he continues with azathioprine, lisinopril and a low prednisone dose without evidence of liver or kidney disease activity.
Key words: Glomerulosclerosis, Focal Segmental, Hepatitis, Autoimmune, Proteinuria
Estudiante de 19 años, previamente sano, que consulta por cuadro de 1 semana de evolución de gonalgia bilateral leve a moderada, sin limitación funcional, factores gatillantes, sintomatología asociada, ni requerimientos de analgesia durante su evolución. Se encontraba en buenas condiciones generales, signos vitales normales y un examen articular que, aparte del dolor, no evidenciaba alteraciones inflamatorias; el resto del examen físico era normal.
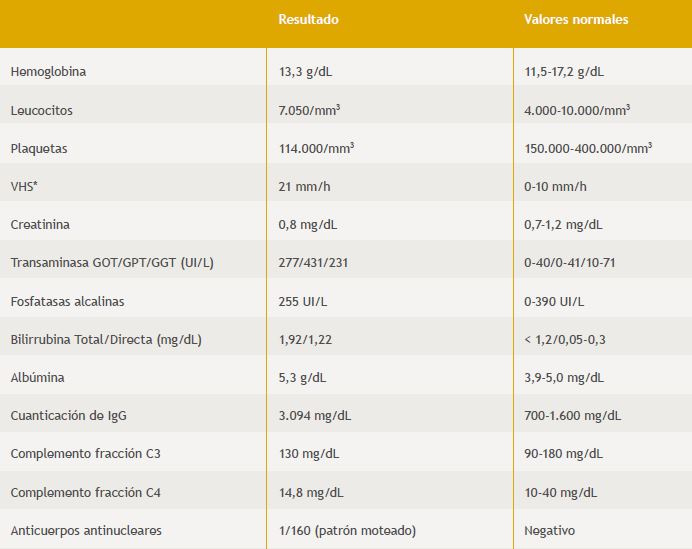
En el estudio inicial (Tabla 1) destacaba hemograma con trombocitopenia leve, serie roja y leucocitos normales y velocidad de sedimentación aumentada. En lo bioquímico, una elevación marcada de transaminasas, hiperbilirrubinemia de predominio directo, leve aumento de γ-glutamil-traspeptidasa (GGT) con fosfatasas alcalinas (FA), albúmina sérica y protrombina normales No se identificó exposición a fármacos o tóxicos en los 6 meses previos y el estudio serológico para virus hepatotropos fue negativo. La ecografía abdominal fue normal, vía biliar fina, sin colelitiasis, con riñones de tamaño y morfología conservados. El examen de orina sólo mostraba proteinuria, que cuantificada resultó en 2,3 g/24 h. No había alteraciones de laboratorio que sugirieran acidosis tubular.
Tabla 1. Exámenes de laboratorio al debut clínico de paciente con Hepatitis autoinmune y glomeruloesclerosis focal y segmentaria.

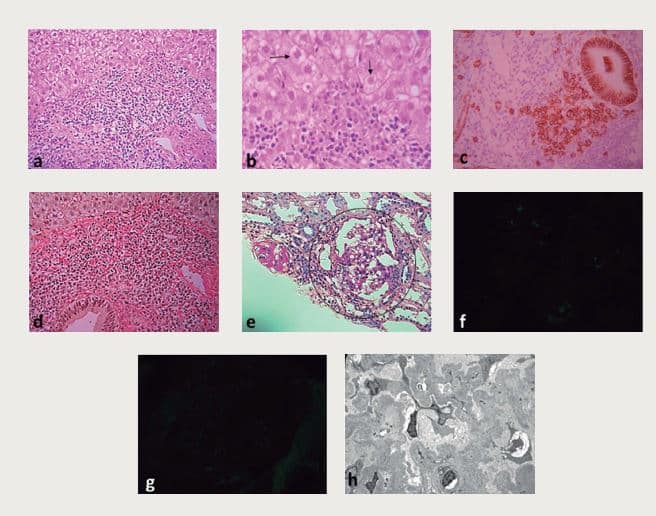
Dadas las manifestaciones multisistémicas (articular, renal y hepático) se sospechó una condición autoinmune, particularmente un debut de Lupus Eritematoso Sistémico (LES), obteniéndose anticuerpos antinucleares (ANA) positivos en 1/160, patrón moteado, pero con Anti-DNA negativo y complemento C3 y C4 normales. Todo ello motivó el estudio de autoinmunidad hepática, destacando la presencia de anticuerpos anti músculo liso (ASMA) positivos e IgG elevada, lo que orientó el diagnóstico hacia una Hepatitis Autoinmune (HAI), que fue confirmada por una biopsia hepática con lesiones características (Figura 1 fila superior). Inició entonces terapia esteroidal oral y azatioprina 100 mg/día logrando normalización de las alteraciones hepáticas y ausencia de sintomatología en tres semanas. Sin embargo, la proteinuria persistió por los siguientes 6 meses, en valores de 1 g/24 h, sin hematuria ni deterioro funcional, realizándose biopsia renal que mostró una Glomeruloesclerosis Focal y Segmentaria variedad NOS (“not otherwise specified”, sin alteraciones específicas ó clásica), con fusión pedicelar completa, ausencia de depósitos inmunes en la inmunofluorescencia y microscopía electrónica y compromiso túbulo-intersticial crónico, discreto y focal (Figura 1, fila inferior). La adición de terapia antiproteinúrica inespecífica (lisinopril oral a 10 mg/día) indujo rápida normalización a 0,2 g/24 h y tras 30 meses se mantiene con mínima dosis de prednisona, 100 mg de azatioprina e igual dosis de lisinopril, sin evidencia de actividad de sus patologías.
Figura 1. Lesiones histológicas. Fila superior (hígado) mostrando (a) infiltrado inflamatorio mononuclear con actividad de interfase (disrupción de la placa limitante); (b) balonización de los hepatocitos (flechas); (c) identificación de plasmocitos de localización periportal (reactividad de membrana celulares en inmunohistoquímica CD 138) y (d) escasa fibrosis (fibras colágenas en rojo) en la tinción de Van Gieson. Fila inferior (riñón): e) microscopía óptica en tinción de ácido peryódico de Schiff (PAS) mostrando la lesión de esclerosis glomerular focal (glomérulo destacado en círculo); (f) y (g) inmunofluorescencia glomerular negativa para C1q e IgG, respectivamente. En (h) microscopía electrónica con plegamiento de la membrana basal glomerular, ausencia de depósitos inmunes y desaparición completa de los pedicelos.
Discusión
Este caso muestra un desafío de diagnóstico diferencial para un cuadro polimorfo, con manifestaciones diversas y potencialmente muy grave en un joven previamente sano. Inicialmente pareció razonable buscar una única entidad como responsable de todas las manifestaciones, la que sí debía explicar todos los mecanismos patogénicos. Por la edad y compromiso multiorgánico, un LES apareció como primera posibilidad, dada su alta frecuencia de compromiso renal. Sin embargo en el LES, si bien la afectación hepática puede ser descrita hasta en 50% de los casos, ella es de causa muy variada y rara vez constituye una condición grave1; en los hechos, el compromiso hepático no forma parte de los criterios diagnósticos de LES del Colegio Americano de Reumatología. Una serie de 248 pacientes con lupus y manifestaciones hepáticas mostró que aunque 15% tenía algún grado de alteración enzimática, sólo 1/3 de ellos tenía una “hepatitis lúpica” propiamente tal2 y más frecuentemente era explicable por toxicidad por drogas, esteatosis hepática, hepatitis virales, presencia de HAI sobreagregada o hepatopatía crónica subyacente3.
El compromiso inicial fue compatible con hepatitis leve (sin compromiso de síntesis), con aumento de transaminasas y discreta elevación de bilirrubina, de predominio directo y sin colestasia, sugiriendo daño hepatocelular4. Consecuentemente, se buscó descartar lo más frecuente4, incluyendo los virus hepatotropos, y realizar estudio ecográfico para definir integridad estructural (cirrosis, esteatosis, etc), descartar un Sindrome de Budd Chiari y comprobar normalidad de vía biliar, pues una litiasis podría ocasionar ascenso de transaminasas de rápida resolución. Adicionalmente descartamos la ingesta exagerada y crónica de alcohol, uso de fármacos o sustancias potencialmente hepatotóxicas4 y desechamos la esteatohepatitis no alcohólica4,5.
La HAI es una enfermedad inflamatoria crónica, caracterizada por manifestaciones inmuno-serológicas que, a principios de los 50’s y por sus hallazgos autoinmunes, fue denominada “hepatitis lúpica o lupoide”6. Afecta mayoritariamente a mujeres (70%), de cualquier edad, siendo más frecuente entre los 40-50 años, sin predilección étnica7. Su prevalencia se ha estimado cercana a 20/100.000 habitantes en Europa, con incidencias variables según país y data del estudio8. En 75% de los casos debuta como hepatitis crónica que, sin tratamiento, puede evolucionar hacia cirrosis y sus complicaciones7. Si bien el diagnóstico se basa habitualmente en la presencia de autoanticuerpos (ANA y/o ASMA), niveles elevados IgG circulante y el descarte de otras hepatopatías, la presencia aislada de autoanticuerpos, incluso en títulos altos, son insuficientes para ello6. Sabidas las coincidencias serológicas entre LES y HAI, existen marcadores específicos para distinguirlas. Los anticuerpos Anti DNA de doble cadena y anti Smith, si bien pueden aparecer en algunas HAI, están mucho más asociados a LES; por otro lado, ASMA y anti Liver Kidney Microsomal constituent (LKM-1) son marcadores muy específicos de HAI, y por lo general no se encuentran en LES9. En el caso presentado, el estudio serológico fue compatible y existió confirmación histológica según criterios establecidos10. La ausencia de anticuerpos anti-LKM-1 y la rápida respuesta a terapia esteroidal e inmunosupresora son su- gerentes de HAI tipo I, la más frecuentemente descrita11. Reconocida la validez de la serología, la histología sigue siendo requisito fundamental para el diagnóstico definitivo y ha sido incluida en scores simples que permiten definir la enfermedad como probable o definitiva, información de gran ayuda terapéutica10. Ella está bien definida para enfermedad activa (como nuestro caso), donde los corticoides sistémicos aislados o combinados con inmunomoduladores constituyen la primera línea para suprimir la actividad inflamatoria a largo plazo y evitar la progresión hacia estadios más avanzados de enfermedad7,11.
La HAI puede tener manifestaciones extrahepáticas hasta en 50% de los pacientes12, como expresión de la enfermedad en otros órganos (frecuentes en piel y articulaciones) las que evolucionan favorablemente con la terapia12. Pueden coexistir otras enfermedades autoinmunes, muy raramente del ámbito renal (< 1% de los casos), habiéndose descrito excepcionalmente glomerulopatía membranosa13 pero nunca la GSFS.
La asociación proteinuria-daño hepatocelular ha sido descrita para glomerulopatías asociadas a infecciones virales hepatotropas, teniendo como sustrato histológico renal la GSFS, como ocurre con los virus de Inmunodeficiencia Humana Tipo 1 (VIH), citomegalovirus, hepatitis B, Epstein-Barr y Parvovirus B19 (los últimos dos no estudiados en nuestro caso). Respecto del citomegalovirus, es característica la lesión glomerular colapsante14, con síndrome nefrótico e importante compromiso funcional renal, lo que no corresponde al cuadro clínico aquí reportado.
La GSFS constituye una glomerulopatía frecuente, que da cuenta de 40% de los síndromes nefróticos del adulto. Más que una enfermedad, es vista actualmente como un grupo de síndromes clínico-patológicos que comparten un patrón histológico común, mediado por una lesión podocitaria de causa variada. En 80% de los casos no tiene causa identificable y es denominada idiopática o primaria, siendo probablemente mediada por factores inmunológicos15. Las otras obedecen a alteraciones genéticas de las proteínas involucradas en el diafragma de filtración glomerular (incluidas proteínas podocitarias), daño adaptativo a insuficiente masa nefronal, u otros agentes como infecciones, drogas, etc. La inmunofluorescencia y microscopía electrónica permiten develar ocasionalente, detrás de un patrón histológico de GSFS, la presencia de nefropatías mediadas por complejos inmunes (IgA, Lupus etc), con pronóstico y tratamiento muy diferentes.

En nuestro paciente, el patrón inespecífico de esclerosis (NOS), sin depósitos inmunes y con fusión pedicelar completa, es sugerente de una GSFS primaria16, no observándose glomerulomegalia o distribución preferentemente perihiliar de la lesión, propias de las formas adaptativas con hiperfiltración como se describe en la obesidad y post-nefrectomía17. Sin perjuicio de lo anterior y aunque no existen antecedentes familiares de nefropatía, debemos destacar que la presentación histológica de una GSFS primaria puede ser indistinguible de aquella producida por las mutaciones genéticas18, estudio que no fue realizado. Adicionalmente, algunas GSFS han sido asociadas al uso de fármacos (litio, sirolimus) o drogas (heroína), algunas de las cuales pueden inducir además hepatoxicidad (anabólicos) las que no son planteables en este caso19.
Es destacable que, pese al adecuado y prolongado control de la actividad de la HAI, persistiera la proteinuria sugiriendo una glomerulopatía independiente. La precisión diagnóstica lograda fue trascendente, evitando los riesgos asociados a la intensa terapia inmunosupresora que hubiera implicado un LES. Un diagnóstico diferencial amplio y sistemático, utilizando todas las herramientas disponibles permitió establecer diagnósticos precisos, oportunos y diseñar una terapia que puede mejorar el pronóstico de ambas patologías, las que por su presentación, evolución clínica, respuestas terapéuticas y hallazgos histológicos, parecen corresponder a dos enfermedades simultáneas, coexistentes. Sin perjuicio de ello, la introducción de estudios genéticos complejos como los “genome-wide association studies” (GWAS)20 podrían, a futuro, esclarecer ésta y otras asociaciones clínicas que hoy carecen de una explicación razonable.
Agradecimientos
Al Dr. Alvaro Ibarra V., Anátomo-Patólogo, Clínica Las Condes, Santiago, por el informe histológico y la colaboración con las imágenes de la biopsia hepática.
Trabajo no recibió financiamiento.
Referencias Bibliográficas
1. De Santis M, Crotti C, Selmi C. Liver abnormalities in connective tissue diseases. Best Pract Res Clin Gastroenterol 2013; 27 (4): 543-51. [Links]
2. Piga M, Vacca A, Porru G, Cauli A, Mathieu A. Liver involvement in systemic lupus erythematosus: incidence, clinical course and outcome of lupus hepatitis. Clin Exp Rheumatol 2010; 28 (4): 504-10. [Links]
3. Grover S, Rastogi A, Singh J, Rajbongshi A, Bihari C. Spectrum of Histomorphologic Findings in Liver in Patients with SLE: A Review. Hepat Res Treat 2014; 2014 562979. [Links]
4. Aithal GP, Watkins PB, Andrade RJ, Larrey D, Molokhia M, Takikawa H, et al. Case definition and phenotype standardization in drug-induced liver injury. Clin Pharmacol Ther 2011; 89 (6): 806-15. [Links]
5. European Association for the Study of the L, European Association for the Study of D, European Association for the Study of O. EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J Hepatol 2016; 64 (6): 1388-402. [Links]
6. Bogdanos DP, Invernizzi P, Mackay IR, Vergani D. Autoimmune liver serology: current diagnostic and clinical challenges. World J Gastroenterol 2008; 14 (21): 3374-87. [Links]
7. Gatselis NK, Zachou K, Koukoulis GK, Dalekos GN. Autoimmune hepatitis, one disease with many faces: etiopathogenetic, clinico-laboratory and histological characteristics. World J Gastroenterol 2015; 21 (1): 60-83. [Links]
8. European Association for the Study of the L. EASL Clinical Practice Guidelines: Autoimmune hepatitis. J Hepatol 2015; 63 (4): 971-1004. [Links]
9. Beisel C, Weiler-Normann C, Teufel A, Lohse AW. Association of autoimmune hepatitis and systemic lupus erythematodes: a case series and review of the literature. World J Gastroenterol 2014; 20 (35): 12662-7. [Links]
10. Hennes EM, Zeniya M, Czaja AJ, Pares A, Dalekos GN, Krawitt EL, et al. Simplified criteria for the diagnosis of autoimmune hepatitis. Hepatology 2008; 48 (1): 169-76. [Links]
11. Mieli-Vergani G, Vergani D, Czaja AJ, Manns MP, Krawitt EL, Vierling JM, et al. Autoimmune hepatitis. Nature Reviews Disease Primers. 2018;4:18017. [Links]
12. Wong GW, Heneghan MA. Association of Extrahepatic Manifestations with Autoimmune Hepatitis. Dig Dis 2015; 33 Suppl 2: 25-35. [Links]
13. Takahashi K, Takasaki S, Morita C, Hayashida K, Matsui M, Tamechika Y, et al. Autoimmune hepatitis with membranous glomerulonephritis. J Gastroenterol Hepatol 2001; 16 (3): 356-9. [Links]
14. Tomlinson L, Boriskin Y, McPhee I, Holwill S, Rice P. Acute cytomegalovirus infection complicated by collapsing glomerulopathy. Nephrol Dial Transplant. 2003; 18 (1): 187-9. [Links]
15. Kronbichler A, Leierer J, Oh J, Meijers B, Shin JI. Immunologic Changes Implicated in the Pathogenesis of Focal Segmental Glomerulosclerosis. Biomed Res Int 2016; 2016: 2150451. [Links]
16. Deegens JK, Dijkman HB, Borm GF, Steenbergen EJ, van den Berg JG, Weening JJ, et al. Podocyte foot process effacement as a diagnostic tool in focal segmental glomerulosclerosis. Kidney Int 2008; 74 (12): 1568-76. [Links]
17. Rosenberg AZ, Kopp JB. Focal Segmental Glomerulosclerosis. Clin J Am Soc Nephrol 2017; 12 (3): 502-17. [Links]
18. Lepori N, Zand L, Sethi S, Fernández-Juárez G, Fervenza FC. Clinical and pathological phenotype of genetic causes of focal segmental glomerulosclerosis in adults. Clin Kidney J 2018; 11 (2): 179-90. [Links]
19. D’Agati VD, Kaskel FJ, Falk RJ. Focal segmental glomerulosclerosis. N Engl J Med 2011; 365 (25): 2398-411. [Links]
20. Webb GJ, Hirschfield GM. Using GWAS to identify genetic predisposition in hepatic autoimmunity. J Autoimmun 2016; 66: 25-39. [Links]
This is an Open Access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, istribution, and reproduction in any medium, provided the original work is properly cited.

Más notas de la edición 95
Lee desde Issuu nuestra última edición publicada en Octubre 2025, Edición número 170

Notas relacionadas a Daño hepatocelular, proteinuria y autoinmunidad:...


