



Complicaciones a Largo Plazo de las Glucogenosis Hepáticas Tipo I, III, VI y IX
Elena Martín Hernández U. Pediátrica de Enfermedades Raras. E. Mitocondriales-Metabólicas Hereditarias Hospital Universitario 12 de Octubre. Madrid.
1.- INTRODUCCIÓN
Hace tan solo unos años, la preocupación de los pediatras con los niños afectos de glucogenosis hepática, en especial con glucogenosis I (GSD I), era que presentaran el menor número posible de descompensaciones con hipoglucemia y acidosis láctica, y que tuvieran un crecimiento y un desarrollo psicomotor normales. Con el paso del tiempo, la mejoría en el tratamiento dietético y las medidas de prevención de las descompensaciones, se ha conseguido que estos niños crezcan y se desarrollen con normalidad, alcanzando la edad adulta la mayoría de ellos, por lo que hoy nuestro interés se centra en tratar de evitar la aparición de complicaciones a largo plazo y, en definitiva, lograr para ellos una expectativa de vida normal.
Debido a que las glucogenosis son enfermedades raras, con una incidencia baja en la población general, y a las características de nuestro sistema sanitario, el número de pacientes por cada centro es escaso, por lo que es difícil para los profesionales adquirir experiencia propia. Por otra parte en la literatura médica hay gran diversidad en el manejo de los pacientes entre unos centros y otros. En 1996 se inició un Estudio Colaborativo Europeo que reunió a pacientes de toda Europa con GSD I, con objeto de analizar las manifestaciones clínicas, complicaciones y los diversos tratamientos utilizados, y sacar unas guías generales de seguimiento y tratamiento que se publicaron en 2002 [1] [2] [3]. Posteriormente no se han hecho otros estudios multicéntricos similares, pero sí hay publicaciones dirigidas a complicaciones concretas con suficiente número de casos, que nos permiten analizar los diferentes problemas que se pueden presentar a lo largo de la vida en estos pacientes así como la manera de prevenirlos, detectarlos y tratarlos.
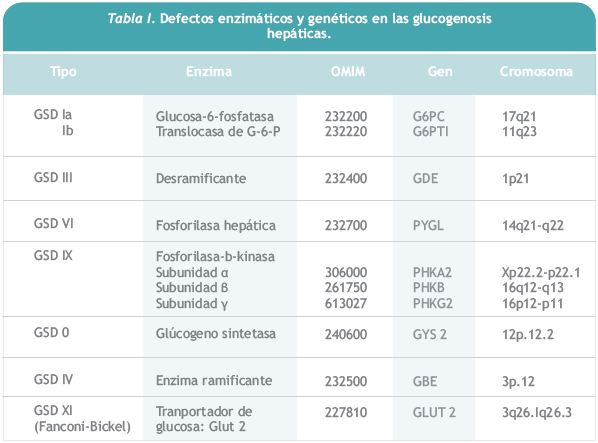
En esta revisión vamos a referirnos a las glucogenosis hepáticas que cursan con hepatomegalia e hipoglucemia en la infancia, o sea, a las GSD I, III, VI y IX ya que hay otras, las GSD 0, IV y el síndrome de Fanconi-Bickel, que difieren en sus manifestaciones clínicas y en sus complicaciones a largo plazo. Los defectos genéticos y enzimáticos de cada una de ellas se encuentran recogidos en la Tabla 1. En las glucogenosis III, VI y IX el defecto enzimático impide la obtención de glucosa a partir del glucógeno almacenado en el hígado (glucogenólisis), pero se puede obtener a partir de otros metabolitos, como aminoácidos, glicerol, etc. (neoglucogénesis) (Figura 1). En la GSD I el defecto enzimático afecta tanto a la glucogenólisis como a la neoglucogénesis, por lo que son las más graves, con debut clínico más precoz y menor tolerancia al ayuno.
2.- GLUCOGENOSIS IA (GSD IA)
La GSD Ia (OMIM 232200) se debe a un defecto en el gen G6PC, que ocasiona un déficit de la actividad enzimática glucosa-6-fosfatasa, que es la enzima encargada de transformar la glucosa 6-fosfato (glucosa 6-P) en glucosa en el retículo endoplásmico rugoso. Así pues, están interrumpidas tanto la vía de la glucogenólisis, paso de glucógeno a glucosa, como de la neoglucogénesis, transformación de otros metabolitos (aminoácidos, glicerol, fructosa) en glucosa, por lo cual se produce hipoglucemia tras ayunos cortos, 2-3 horas tras la ingesta. El organismo, en un intento de normalizar la glucemia, activa la glucogenólisis y neoglucogénesis, pero con ello lo único que se consigue es un acúmulo de glucosa 6-P que se desvía hacia otras vías: síntesis de glucógeno que se acumula en el hígado; vida de las pentosas que termina con la producción de ácido úrico, y vía de la glucolisis anaerobia con aumento de ácido láctico, acetil CoA y síntesis de ácidos grasos y triglicéridos (Figura 1).
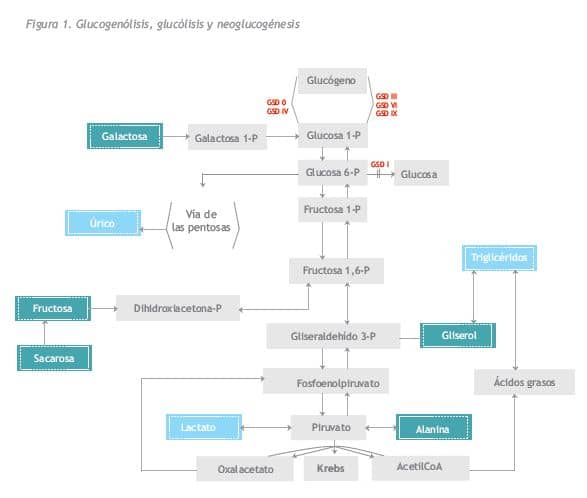
La glucosa 6 fosfatasa se expresa en hígado, riñón e intestino y es en estos órganos, fundamentalmente en los dos primeros, donde se acumula el glucógeno.
2.1.- Complicaciones hepáticas
La hepatomegalia, disminuye al inicio del tratamiento, persistiendo no obstante a lo largo de la evolución; no se acompaña de esplenomegalia.
Se debe al depósito de glucógeno y a la infiltración grasa. Los pacientes con GSD Ia no suelen evolucionar a cirrosis y fallo hepático, presentan hipertransaminasemia sin alteración de otros parámetros de función hepática. Las complicaciones hepáticas más frecuentes son los adenomas, tumores benignos cuya causa se desconoce [4] [5].
Aparecen en la adolescencia, estando presentes en un 70% de los pacientes mayores de 25 años. Suelen ser pequeños, múltiples y mal encapsulados. En ocasiones pueden sangrar, ocasionando dolor y anemia, y otras veces pueden comprimir otras estructuras o romperse. El crecimiento es lento, pero en un 10% de los casos pueden malignizarse.
El diagnóstico es por ecografía y en caso de sospecha de malignización se realiza resonancia magnética. El valor de las alfafetoproteínas en estos casos está cuestionado.
El tratamiento es la resección o el trasplante hepático.
La prevención de las complicaciones hepáticas es consiguiendo un excelente control metabólico [6] [7], y evitando el alcohol y fármacos hepatotóxicos, en especial algunos contraceptivos orales.
El trasplante hepático, hoy prácticamente está reservado para casos de adenomas múltiples con sospecha de malignización. No se indica para obtener un mejor control metabólico, ya que en los pacientes en que el cumplimiento dietético es malo también lo será el cumplimiento del tratamiento inmunosupresor con riesgo para el injerto y para la vida del paciente. Además, con el trasplante no se evitan las complicaciones extrahepáticas [8].
2.2.- Complicaciones renales
La nefromegalia, consecuencia del depósito de glucógeno, suele estar presente en el momento del diagnóstico y persiste a lo largo de la evolución. En algunas ocasiones hay también una tubulopatía proximal que desaparece al iniciarse el tratamiento y conseguirse el control metabólico. Las complicaciones a largo plazo son parecidas a las de la nefropatía diabética: hiperfiltración, microalbuminuria, proteinuria, hipertensión arterial y fallo renal. En un estudio reciente efectuado en 39 pacientes de una edad media de 11,6 años (rango 0,8-23) presentaron hiperfiltración el 67% de los casos, microalbuminuria y proteinuria el 67 % y 40% respectivamente de los que tenían más de 18
años, hipertensión arterial el 5,12% y todos tenía valores normales de urea y creatinina, aunque hay que señalar que eran pacientes muy jóvenes [9]. En otras series se ha comunicado hipertensión arterial entre el 7 y el 23% de los casos, e insuficiencia renal en el 2%. Los autores señalan que estas complicaciones pueden retrasarse con un buen control metabólico y con la prescripción de inhibidores del enzima convertidora de la angiotensina (IECA) cuando se demuestre microalbuminuria durante más de 3 meses. Afectación tubular distal, con hipercalciuria e hipocitraturia, puede aparecer en algunos pacientes a pesar del buen control metabólico; en estos casos la alcalinización de la orina con citrato potásico previene la aparición o progresión de nefrocalcinosis y urolitiasis.
Con respecto a las complicaciones urológicas, en un estudio de este mismo año se ha demostrado urolitiasis en un 65% de los pacientes, versus un 5% en la población general. Los factores predisponentes a esta complicación son la hipercalciuria, hiperuricosuria, hiperoxaluria y sobre todo la hipocitraturia. Como ya dijimos antes se pueden prevenir con la administración de citato potásico en caso de hipercalciuria e hipocitraturia y con alopurinol en caso de hiperuricemia [10].
Para la detección de las complicaciones renales se recomienda realizar determinación de microalbuminuria y proteinuria una vez al año en menores de 6 años y a partir de entonces cada 6 meses. Estudios de función renal completos se realizarán anualmente y ecografía renal coincidiendo con los controles hepáticos.
2.3.- Complicaciones osteoarticulares
La mayor expectativa de vida en los pacientes con glucogenosis I, hace que aparezcan otras complicaciones, como la osteoporosis, que hace unos años no nos preocupaban, de hecho en el estudio colaborativo europeo no se hace referencia a ella. Hay muchos factores que predisponen a la osteoporosis: reducción en la glicosilación de las proteínas de la matriz ósea, déficit de insulina y exceso de glucocorticoides, disminución de la masa muscular y de la actividad física, defecto en la mineralización en relación con alteraciones metabólicas como acidosis o pérdida de calcio, fósforo y magnesio, y por último a deficiencia en vitamina D, ya sea por una dieta restrictiva o por una disminución en la absorción a nivel intestinal [11] [12] [13] . En un estudio realizado en 29 pacientes con GSD Ia de diferentes edades se observó en la densitometría, que el z-score (mineralización) de los casos prepuberales era normal, mientras en los adolescentes y adultos estaba disminuido[11].
En lo referente a la talla final, en el estudio colaborativo europeo en 288 pacientes nacidos entre 1943 y 1996 , se encontraba por debajo de 2DS en el 35% de los casos con GSD Ia y en el 50% de los casos con GSD Ib. Hay que tener en cuenta que en esta serie se recogen casos antiguos en los que no se disponía del tratamiento actual. En otro estudio más reciente en 30 pacientes con GSD I entre 0,8 y 33 años la talla media fue –1,6 DS, mejor que en una serie anterior en la misma institución [12]. La mayoría de nuestros casos actuales en edad pediátrica tienen una talla normal. La reducción en la talla final se ha atribuido a factores hormonales, como una menor sensibilidad a la hormona del crecimiento, un exceso de glucocorticoides y a los menores valores de insulina ante la sobrecarga de insulina.
La gota, otra de las complicaciones osteoarticulares es consecuencia de la hiperuricemia pero es poco frecuente. En el estudio europeo se refiere una frecuencia de urolitiasis o gota en el 14% de los casos con hiperuricemia mantenida.
La detección de las complicaciones osteoarticulares se hace mediante calibraciones seriadas de la dieta, seguimiento de la velocidad de crecimiento y determinaciones periódicas de calcio, fósforo, fosfatasa alcalina, ácido úrico y vitamina D. Asimismo se realizarán densitometrías en función de los datos clínicos y analíticos. Si los aportes dietéticos de calcio y vitamina D son deficientes o hay alteraciones analíticas habrá que dar suplementos. En todos los casos se animará a hacer ejercicio físico. En los casos de hiperuricemia se recomendará tratamiento con alopurinol.

2.4.- Complicaciones cardiovasculares y pulmonares
A pesar de presentar factores que incrementan el riesgo de aterogénesis, como hipertrigliceridemia (70%), hipercolesterolemia (40%), descenso del HDL colesterol y disfunción endotelial, no hay evidencia clínica de una mayor presencia de complicaciones cardiovasculares en estos pacientes. En el estudio europeo se comunicó hipertensión en el 7% de los pacientes y solo se observó aterogénesis en una paciente que falleció tras su segundo trasplante renal. Cardiopatía isquémica se ha comunicado entre el 0 y el 6% según diferentes series. Recientemente se ha publicado un trabajo en el que se demuestra un engrosamiento de la capa íntima de la carótida y un aumento de la tonometría en la arteria radial, ambos factores indicativos de afectación vascular [14].
Para conocer con más precisión el alcance de estas complicaciones habrá que esperar a que los pacientes vayan alcanzando edades más avanzadas.
Para la detección y prevención de las complicaciones cardiovasculares habrá que tomar periódicamente la tensión arterial y hacer determinaciones seriadas de colesterol y triglicéridos. En adultos se recomienda hacer ecocardiograma una vez al año. Se tratará de mantener los valores de triglicéridos y colesterol dentro de los límites indicados para estos pacientes mediante la dieta, y si con ello no es posible se añadirán estatinas o fibratos. En caso de hipertensión se tratará con inhibidores de la enzima convertidora de angiotensina. Es muy importante mentalizar a los pacientes desde la niñez de lo nocivo que es el tabaco.
La hipertensión pulmonar es una complicación infrecuente, pero grave cuando aparece. Se suele presentar en la segunda o tercera década de la vida y en general en casos con mal control metabólico. Se detecta en los controles ecocardiográficos con una elevación en la presión del ventrículo derecho. Clínicamente se manifiesta por fatigabilidad y dificultad respiratoria con el ejercicio, llegando a ocasionar insuficiencia cardiaca y cuadros sincopales. El pronóstico es malo, aunque se han comunicado buenos resultados al tratamiento con beraprost, sildenafilo y bosentan [15].
2.5.- Complicaciones digestivas
Pueden aparecer pancreatitis y colecistitis en relación con hipertrigliceridemia y tendrán que descartarse en casos de dolor abdominal agudo con los estudios necesarios. Para su prevención es necesario mantener los triglicéridos dentro de la normalidad con la dieta o, si es necesario, mediante el tratamiento con fibratos.

La diarrea es frecuente (33%) aunque en menor grado que en los casos de GSD Ib1. Se debe a los depósitos de glucógeno en la pared intestinal.
2.6.- Complicaciones neurológicas
El pronóstico neurológico en general es bueno. En el estudio colaborativo europeo se refiere que un 89% de los niños seguían una escolarización normal y un 84% de los adultos, o seguían sus estudios, o estaban trabajando. El CI era inferior a 85 en 16% de los casos. De nuevo hay que tener en cuenta que es un estudio con pacientes antiguos.
En la actualidad, nuestros pacientes en edad pediátrica tienen por lo general un desarrollo psicomotor normal.
2.7.- Complicaciones ginecológicas y en la gestación
Este tipo de complicaciones no se han estudiado en profundidad. La pubertad se encuentra retrasada aproximadamente en un 50% de los casos y se han observado quistes ováricos en 3 de 7 pacientes a las que se realizó ecografía pélvica.
En los últimos años están apareciendo publicaciones de embarazos en pacientes con GSD Ia. En una serie de 15 gestaciones en 11 pacientes, los resultados fueron buenos salvo en un caso que presentó hiperlactacidemia y deterioro multiorgánico durante el parto con importantes secuelas en el recién nacido. En el resto de los casos el embarazo se desarrolló sin incidencias importantes, hubo un incremento en las necesidades de glucosa durante el primer trimestre y dos partos prematuros de 34 y 35 semanas. En cuanto a repercusiones para la madre, se observó cierto deterioro de la función renal en pacientes con microalbuminuria previa y solo apareció un adenoma nuevo en un caso.

Los autores recomiendan planificar la gestación, retirar alopurinol e IECA en caso de que se estuvieran tomando, extremar los controles de glucemia durante el primer trimestre, evitar la cetosis y administrar una perfusión de glucosa durante el parto y hasta que se normalice la ingesta [16].
3.- GLUCOGENOSIS IB (GSD IB)
En la GSD Ib (OMIM 232220) el enzima deficiente es la translocasa de glucosa-6-fosfato que es la encargada de transportar la glucosa-6-fosfato al interior del retículo endoplásmico rugoso, donde se encuentra la glucosa-6-fosfatasa que la convierte en glucosa.
Estos pacientes presentan las mismas complicaciones que los casos con GSD Ia y además tienen problemas hematológicos, digestivos y tiroideos. Las guías para el seguimiento y tratamiento se extrajeron del estudio colaborativo europeo, al que nos hemos venido refiriendo [3].
3.1.- Complicaciones hematológicas y digestivas
En la médula ósea la glucosa-6-fosfato interviene en la síntesis de productos antioxidantes como el NADPH y el glutatión, que protegen a los neutrófilos de los radicales libres. Cuando están deficientes aparece neutropenia y alteraciones en la función de los mismos, aumentando el riesgo de presentar infecciones y enfermedad inflamatoria intestinal, lo que empeora el pronóstico. La mortalidad en el estudio europeo fue del 12% en estos casos frente a 3,8% en los de GSD Ia. Desde la década de los 90 se han venido utilizando perfusiones de factores estimulantes de colonias de granulocitos (G-CSF) que han mejorado los resultados. Las indicaciones son:neutropenia inferior a 200 x 106/L; infecciones que hayan requerido antibioterapia intravenosa; enfermedad inflamatoria intestinal constatada por ileocolonoscopía, y diarrea que haya requerido ingreso hospitalario o impedido una vida normal [3]. En un estudio reciente llevado a cabo en 7 pacientes entre 5 y 29 años se demostró que la administración de vitamina E a dosis de 600 mg al día en niños y 900 mg al día en adultos disminuía los procesos infecciosos y mejoraba los valores de neutrófilos totales y las lesiones intestinales en la ileocolonoscopía [17].
3.2.- Complicaciones tiroideas
Se ha visto que el 57% de los pacientes con GSD Ib presentan hipotiroidismo. La causa exacta no se conoce, puede deberse a los depósitos de glucógeno en tiroides, a amiloidosis en relación con las infecciones o a fenómenos de autoinmunidad. Para detectarlo hay que realizar estudios de función tiroidea de manera seriada y el tratamiento es con hormonas tiroideas [18].

3.3.- Complicaciones ginecológicas
Recientemente se ha publicado la primera serie de 3 pacientes con GSD Ib y 5 embarazos. Las 3 eran casos leves, ya que ninguna tenía enfermedad inflamatoria intestinal, adenomas hepáticos, ni requería tratamiento con G-CSF. Durante la gestación se incrementaron las necesidades de glucosa, sobre todo durante el segundo trimestre, y las tres presentaron neutropenia, en dos de ellas aparecieron úlceras orales y en la tercera infección urinaria, pero ninguna requirió G-CSF. Los recién nacidos no presentaron problemas significativos. Los autores recomiendan ajustar la ingesta para mantener siempre niveles de glucosa superiores a 75 mg/dL y láctico < 2 mmol/L; administrar suero glucosado durante el parto y hasta que la ingesta se normalice; en caso de enfermedad inflamatoria recomiendan continuar con los GCSF y el 5-aminosalícilico (5-ASA) y no utilizar corticoides ni otros inmunomoduladores. En caso de necesitar tratamiento con anticuerpos monoclonales usar el Adalimumad, que es menos inmunogénico [19] [20].
4.- GLUCOGENOSIS III (GSD III)
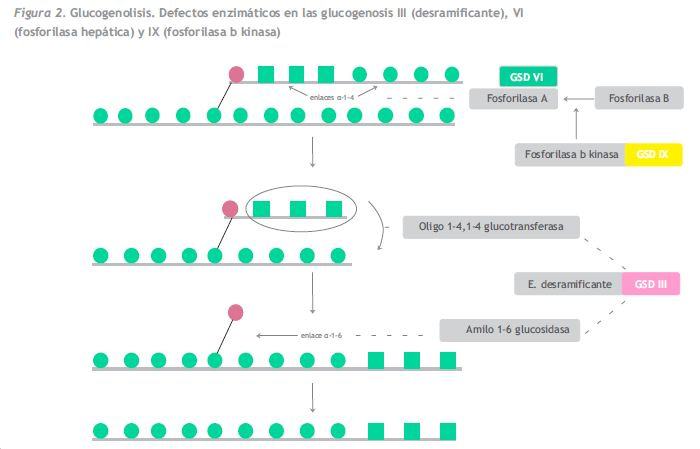
En la GSD III (OMIM 232400) el defecto en el gen GDE, ocasiona una actividad deficiente en la enzima desramificante, responsable de la ruptura de los enlaces alfa-1-6 de la molécula de glucógeno (Figura 2). En situaciones de ayuno se pone en marcha la glucogenólisis o sea, el catabolismo del glucógeno para obtener glucosa, primero actúa la fosforilasa hepática, que va liberando moléculas de glucosa de las cadenas lineales de glucógeno, unidas por enlaces alfa 1-4, y luego actúa el enzima desramificante que rompe los enlaces alfa 1-6 y por tanto las ramificaciones. En caso de deficiencia en esta enzima se inicia la glucogenólisis pero no se termina, acumulándose moléculas de glucógeno anormal, dextrinas, en el hígado y en el músculo en el tipo IIIa (85% de los casos), o solo en el hígado, en el tipo IIIb (15% de los casos). Ambas son producidas por defectos en el mismo gen, habiéndose descrito mutaciones específicas para el tipo b.
Al estar indemne la vía de la neoglucogénesis se puede además obtener glucosa a partir de otros metabolitos como los aminoácidos que proceden del catabolismo proteico y los ácidos grasos que proceden del catabolismo de las grasas. La consecuencia de todo ello, es que la hipoglucemia aparece tras ayunos más prolongados que en la GSD I, que generalmente se asociada a cetosis (por el catabolismo de las grasas), y que la proteinólisis contribuye a la aparición de debilidad muscular.
Las complicaciones a largo plazo de la GSD III son hepáticas, musculares, cardiológicas y óseas [21].
4.1.- Complicaciones hepáticas
La enfermedad hepática suele ser autolimitada y va mejorando con el paso de los años, aunque en algunos casos puede evolucionar a cirrosis y fallo hepático. En la analítica, la hipoglucemia con cetosis aparece tras ayunos más prolongados, la hiperlactacidemia es postprandial y las transaminasas son más elevadas que en la GSDI. La presencia de fibrosis en la biopsia hepática es frecuente en los niños, pero la progresión a cirrosis e insuficiencia hepática es rara. Los adenomas son menos frecuentes que en la GSD I, habiéndose descrito en el 4-20% de los casos, y no se suelen malignizar. En alguna ocasión se han desarrollado hepatocarcinomas en adultos, pero no sobre adenomas, sino sobre lesiones cirróticas. El trasplante hepático está indicado cuando hay fallo hepático o sospecha de malignización, pero no previene la afectación muscular.

4.2.- Complicaciones cardiovasculares y musculares
La miocardiopatía hipertrófica aparece durante la infancia, aunque raramente antes del primer año, en los pacientes con el tipo IIIa. El significado clínico es incierto. Se ha demostrado que con dietas hiperproteicas, con un 20-30% del valor calórico total procedente de las proteínas, se previene o se enlentece su progresión [22].
La miopatía es poco frecuente en niños, manifestándose entre la 3a y 4 adécada de la vida con debilidad muscular y atrofia sobre todo a nivel proximal. Empeora mucho la calidad de vida de estos pacientes. Se puede detener la progresión con las dietas hiperproteicas.
Para el diagnóstico de estas complicaciones realizaremos ecocardiogramas y determinaciones de creatinfosfoquinasa (CPK) seriadas desde la infancia.
La hiperlipemia es menos frecuente que en las GSD I y no hay evidencia clínica de mayor riesgo cardiovascular.
4.3.- Complicaciones óseas
En un estudio reciente llevado a cabo en 15 pacientes con glucogenosis III (12a y 3b), entre 10 y 34 años, el 65% mostraba un densidad ósea inferior a 2DS, o sea, que el riesgo de padecer osteoporosis es alto en estos pacientes. El diagnóstico se hace por densitometría ósea. Los autores lo relacionan con factores musculares, metabólicos y nutricionales y recomiendan un buen control metabólico, calibraciones dietéticas seriadas, dieta rica en proteínas, cubriendo las necesidades en minerales y vitaminas, suplementos de calcio y vitamina D y ejercicio físico regular [23].
5.- GLUCOGENOSIS VI Y IX
Las glucogenosis VI (OMIM 232700) y IX (OMIM 306000, 261750 y 613027), son las más benignas, se expresan en hígado, pero no en el músculo cardiaco o esquelético ni en riñón. Durante la infancia cursan con hipoglucemia, hepatomegalia y retraso del crecimiento, que van mejorando con el paso del tiempo, manteniéndose asintomáticos en la vida adulta. No obstante es prudente hacer seguimiento de la función hepática y de la densidad ósea así como controlar estrechamente la glucemia durante el embarazo [24].
En la Tabla 2 mostramos una serie de recomendaciones generales para evitar la aparición de complicaciones a largo plazo en los pacientes con GSD I, III, VI y IX.

En conclusión, el pronóstico y la calidad de vida en los pacientes con glucogenosis hepáticas, han mejorado enormemente en las últimas décadas con la nutrición enteral a débito continuo, el almidón crudo de maíz, las nuevas harinas modificadas, los G-CSF, y el resto de tratamientos aquí comentados, y sin duda seguirá mejorando en los próximos años, hasta que los nuevos tratamientos en investigación como las transfusiones de hepatocitos y la terapia génica nos permitan obtener la curación definitiva [25] [26].
Referencias Bibliográficas
1. Rake JP, Visser G, Labrune P, Leonard JV, Ullrich K, Smit GPA. Glycogen storage disease type I: diagnosis, management, clinical course and outcome. Results of the European study on glycogen storage disease type I (ESGSD I). Eur J Pediatr 2002; 161:S20-S34.
2. Rake JP, Visser G, Labrune P, Leonard JV, Ullrich K, Smit GPA. Guidelines for management of glycogen storage disease type I- European Study on glycogen storage disease type I (ESGSD I). Eur J Pediatr 2002; 161: S112-S119.
3. Visser G, Rake JP, , Labrune P, Leonard JV, Moses S, Ullrich K, Wendel U, Smit GPA. Consensus guidelines for management of glycogen storage disease type Ib- European study on glycogen storage disease type 1. Eur J Pediatr 2002; 161: S120-S123.
4. Lee JF. Glycogen storage disease type I: pathophysiology of liver adenomas. Eur J Pediatr 2002; 161: S46-S49.
5. Di Rocco M, Calevo MG, Taro M, Melis D, Allegri AEM, Parenti G. Hepatocellular adenoma and metabolic balance in patients with type Ia glycogen storage disease. Mol Genet Metab 2008; 93: 398-402.
6. Daublin G, Schwahn B, Wendel U. Type I glycogen storage disease. Favourable outcome on a strict management regimen avoiding increased lactate production during childhood and adolescence. Eur J Pediatr 2002; 161: S40-S45.
7. Weinstein DA, Wolfsdorf JI. Effect of continuous glucose therapy with uncooked cornstarch on the longterm clinical course of type Ia glycogen storage disease. Eur J Pediatr 2002; 161: S35-S39.
8. Davis MK, Weinstein DA. Liver transplantation in children with glycogen storage disease: controversies and evaluation of the risk/benefit of this procedure. Pediatr Transplantation 2008; 12: 137-145.
9. Martens DHJ, Rake JP, Navis G, Fidler V, van Dael KML, Smit GPA. Renal function in glycogen storage disease type I, natural course, and renopropreservative effects of ACE inhibition. Clin J Am Soc Nephrol 2009; 4: 1741-1746.
10. Scales CD, Chandrashekar AS, Robinson MR, Cantor DA, Sullivan J, Haleblian GE et al. Stones forming risk factors in patients with GSD I. J Urol 2010; 183: 1022-1025.
11. Rake JP, Visser G, Huismans D, Huitema S, van der Veer E, Piers DA. Bone mineral density in children, adolescents and adults with glycogen storage disease type Ia: a crossectional and longitudinal study. J Inherit Metab Dis 2003; 26: 371-384.
12. Mundy HR, Hindmarsh PC, Matthews DR, Leonard JV, Lee PJ. The regulation of growth in glycogen storage disease type 1. Clin Endocrinol 2003; 58: 332-339.
13. Banugaria SG, Austin SL, Boney A, Weber TJ, Kishnani PS. Hypovitaminosis D in glycogen storage disease type I. Mol Gen Metab 2010; 99:434-437.
14. Berner AV, Correia CE, Haller MJ, Theriaque DW, Shuster JJ, Weinstein DA. Vascular dysfunction in glycogen storage disease type I. J Pediatr 2009; 154:588- 91.
15. Ueno M, Murakami T, Takeda A, Kubota M. Efficacy of oral sildenafil in a beraprost-treated patient with severe pulmonary hipertension secondary to type I glycogen storage disease. Circ 2009; 73: 1965-1968.
16. Martens DHJ, Rake JP, Schwarz M, Ullrich K, Weinstein DA, Merkel M, Sauer PJJ, Smit GPA. Pregnancies in glycogen storage disease type Ia. Am J Obstet Gynecol 2008; 198:646.e1-646.e7.
17. Melis D, Della Casa R, Parini R, Rigoldi M, Cacciapuoti C, Marcolongo P, Benedetti A, Gaudieri V, Andria G, Parenti G. Vitamin E supplementation improves neutropenia and reduces the frequency of infections in patients with glycogen storage disease type 1b. Eur J Ped 2009; 168: 1069-1074.
18. Mellis D, Pivonello R, Parenti G, Della Casa R, Salerno M, Lombardi G, Sebastio G, Colao A, Andria G. Increases prevalence of thyroid autoimmunity in patients with glycogen storage disease type I. J Pediatr 2007; 150:300- 305, 305.e1.
19. Dagli AI, Lee PJ, Correia CE, Rodriguez C, Bhattacharya K, Steinkrauss L, Stanley CA, Weinstein DA. Pregnancy in glycogen storage disease type Ib: gestacional care and report of first successful deliveries. J Inherit Metab Dis 2010 (en prensa).
20. Ozen Hasan. Glycogen storage diseases: new perspectives. World J Gastroenterol 2007; 13: 2541-2553.
21. Dagli A, Sentner CP, Weinstein DA. Glycogen storage disease tipe III. In Pagon RA, Bird TC, Dolan CR, Stephens K, editors. GeneReviews [Internet]. Seattle (WA):University of Washington, Seattle; 1993-2010 Mar 9.
22. Dagli AI, Zori RT, McCune H, Ivsic T, Maisenbacher MK, Weinstein DA. Reversal of glycogen storage disease type 3a-related cardiomyopathy with modification of diet. J Inherit Metab Dis 2009.
23. Mundy HR, Willians JE, Lee PJ, Fewtrell MS. Reduction in bone mineral density in glycogenosis type III may be due to a mixed muscle and bone deficit. J Inherit Metab Dis 2008; 31: 418-423.
24. Dagli AI, Weinstein DA. Glycogen storage disease type VI. In Pagon RA, Bird TC, Dolan CR, Stephens K, editors. GeneReviews [Internet]. Seattle (WA): University of Washington, Seattle; 1993-.2009 Apr 23.
25. Koeberl DD, Kishnani PS, Bali D, Chen Y-T. Emerging therapies for glycogen storage disease type I. Trends Endocrinol Metab 2009; 20: 252-258.
Más notas de la edición 14

Lee desde Issuu nuestra última edición publicada en Julio 2024, Edición número 155


Notas relacionadas a Complicaciones a Largo Plazo de las Glucogenosis...
