


Valoración ética de las nuevas opciones reproductivas en las enfermedades mitocondriales
Graciela Moya (1)
1 Instituto de Bioética, Pontificia Universidad Católica Argentina, Argentina
Correspondencia: gramoya@gmail.com
http://dx.doi.org/10.4067/S1726-569X2016000200008
Acta bioeth. vol.22 no.2 Santiago nov. 2016
Diagonal Paraguay #265, Torre 15, Piso 8
Santiago – Chile
Teléfono: (56-2) 29782274
Fax: (56-2) 29782254
info@actabioethica.cl
Resumen
Las enfermedades mitocondriales son un grupo de desórdenes clínicamente heterogéneo con manifestaciones clínicas muy variables y alta tasa de morbi-mortalidad. Tienen complejas implicancias en la reproducción, ya que los riesgos de repetición en la familia son variables, 25% en las formas autosómicas recesivas, alrededor de 4% en los casos de mutaciones mitocondriales de novo, y un riesgo incierto pero elevado si la madre es portadora de las mutaciones mitocondriales. Por ello, se proponen distintas técnicas de diagnóstico preimplantacional, prenatal o de reproducción asistida a fin de evitar el nacimiento de niños con esta patología. En el presente trabajo se describen y analizan las implicancias científicas y éticas de las nuevas técnicas de reproducción asistida que se proponen a las familias con alto riesgo.
Palabras clave: enfermedades mitocondriales, reproducción asistida, trasplante mitocondrial, dignidad humana.
Abstract
Mitochondrial diseases are a clinically heterogeneous group of disorders with variable clinical features and high morbidity and mortality. As there are different hereditary patterns, complex implications in reproduction are expected; the recurrence risk may be about 25% in autosomal recessive inheritance, 4% in de novo mitochondrial mutations, and an uncertain, but high risk, when the mother is carrier of mitochondrial mutations. Thus, different preimplantation and prenatal diagnosis or assisted reproductive techniques are proposed to avoid the birth of children with these diseases. In this paper scientific and ethical implications of new assisted reproductive techniques offer to high risk families are describe and analyzed.
Key words: mitochondrial disorders, assisted reproductive techniques, mitochondrial transplant, human dignity
Resumo
As enfermidades mitocondriais são um grupo de desordens clinicamente heterogêneos com manifestações clínicas muito variáveis e alta taxa de morbi-mortalidade. Têm complexas implicações na reprodução, já que os riscos de repetição na família são variáveis, 25% nas formas autossômicas recessivas, ao redor de 4% nos casos de mutações mitocondriais de novo, e um risco incerto porém elevado se a mãe é portadora das mutações mitocondriais. Por isso, se propõem distintas técnicas de diagnóstico préimplantacional, pré-natal ou de reprodução assistida a fim de evitar o nascimento de crianças com esta patologia. No presente trabalho se descrevem e analisam as implicações científicas e éticas das novas técnicas de reprodução assistida que se propõem às famílias com alto risco.
Palavras-chave: enfermidades mitocondriais, reprodução assistida, transplante mitocondrial, dignidade humana
Introducción
Los avances en el conocimiento del genoma humano, tanto nuclear como mitocondrial, y la aplicación de las nuevas tecnologías reproductivas suscitan un desafío en la aplicación de ambos conocimientos para ampliar las decisiones reproductivas en familias de alto riesgo.
En el presente trabajo se describen y analizan las implicancias científicas y éticas de las nuevas técnicas de reproducción asistida, particularmente el denominado “trasplante mitocondrial”.
Enfermedades mitocondriales
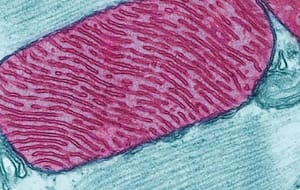
Las enfermedades mitocondriales son un grupo de desórdenes clínicamente heterogéneo, que resulta de la alteración de la función de la cadena respiratoria mitocondrial, esencial en el suministro de energía en forma de ATP. Las proteínas que componen la cadena respiratoria mitocondrial están codificadas por el ADN nuclear o mitocondrial y las proteínas que requiere la mitocondria para su funcionamiento y replicación están codificadas por los genes nucleares. Por ello, las enfermedades mitocondriales pueden originarse a partir de mutaciones del ADN nuclear o del ADN mitocondrial, habiéndose identificado más de 100 loci en la actualidad(1-4).
El ADN mitocondrial (ADNMt) tiene su propio código genético y contiene 37 genes que codifican algunas de las proteínas que intervienen en la cadena respiratoria, y en la síntesis del ARN ribosomal y ARN de transferencia necesarios para la síntesis proteica en las mitocondrias(5). Los individuos normales tienen homoplasmía en sus mitocondrias, es decir, todas las mitocondrias tienen la misma información genética normal. Sin embargo, las mutaciones del genoma mitocondrial pueden estar presentes en todas las mitocondrias, siendo homoplásmicas, o estar presentes en algunas mitocondrias entre otras normales, siendo heteroplásmicas. La mayoría de los pacientes con enfermedades mitocondriales tienen heteroplasmía, con distinto porcentaje de mitocondrias normales y anormales en sus tejidos. Por tanto, la variabilidad en la expresión clínica dependerá del porcentaje de mitocondrias mutadas. Se considera que las manifestaciones clínicas se detectan con un porcentaje de mitocondrias mutadas mayor del 70%(6).
La expresión clínica de las enfermedades mitocondriales es muy variable y puede presentarse en cualquier edad de la vida(7), afectando habitualmente aquellos tejidos que tienen alta demanda metabólica(8). Algunos de estos cuadros pueden afectar a un solo órgano (como la Neuropatía Hereditaria de Leber —LHON—, que afecta la función del nervio óptico) o tener un efecto multisistémico; entre ellos, síntomas neurológicos y miopáticos, que generan alta morbilidad y mortalidad; frecuentemente se presenta ptosis, oftalmoplegía externa, miopatía proximal e intolerancia al ejercicio, cardiomiopatía, sordera sensorineural, atrofia óptica, retinopatía pigmentaria, falla hepática y diabetes mellitus. Los hallazgos de sistema nervioso central son muy variados: encefalopatía, convulsiones, demencia, migraña, episodios tipo derrame cerebral, ataxia y espasticidad(9). Los cuadros clínicos pueden ser más leves, como es el caso de la Oftalmoplegía Externa Crónica Progresiva (CPEO), el Síndrome de Kearns-Sayre (KSS), que asocia a la oftalmoplegía retinopatía pigmentaria, o cuadros más complejos, como la Encefalomiopatía Mitocondrial con Acidosis Láctica y episodios tipo derrame cerebral (MELAS), la epilepsia mioclónica con fibras rojas rasgadas (MERRF), la debilidad neurogénica con ataxia y retinitis pigmentaria (NARP), o el Síndrome de Leigh encefalomielopatía necrotizante subaguda (LS). Los diagnósticos son complejos, dado que las manifestaciones clínicas son muy variables y los pacientes no siempre se pueden clasificar en un único cuadro, porque los síntomas se superponen.
Existe además una mayor incidencia de fallas reproductivas de segundo y tercer trimestre(7). La alteración de la función mitocondrial también se ha descrito asociada con otras enfermedades, ya que existe toda una compleja vía de comunicación entre la mitocondria y otros compartimentos celulares, entre ellas enfermedad de Alzheimer, Parkinson, Huntington, cáncer, enfermedad cardíaca, diabetes, epilepsia, obesidad, trastornos de personalidad, enfermedad psiquiátrica o el envejecimiento. También se ha demostrado que interviene en el metabolismo de toxinas, fármacos, anestésicos, antibióticos, o quimioterápicos(9).
En forma aislada son enfermedades poco frecuentes, pero en su conjunto se observan en uno cada 5.000 a uno cada 17.000 individuos(10,11), incluyendo aquellas debidas a mutaciones tanto del genoma nuclear como del mitocondrial.
El diagnóstico de las enfermedades mitocondriales es clínico, se basa en las manifestaciones clínicas, la historia familiar y los estudios complementarios, y se confirma con estudios de genética molecular del ADN nuclear o mitocondrial.
La herencia es variada. Si la mutación se encuentra en el ADN nuclear, se hereda con un patrón mendeliano autosómico recesivo o dominante, o ligado al X, y cuando afecta al ADNMt se hereda con un patrón mitocondrial o materno, también puede presentarse en forma esporádica, como mutaciones mitocondriales de novo. Por ello, el estudio molecular es complejo; aun hay cuadros como la oftalmoplegía externa, que puede deberse a mutaciones en el ADN nuclear o en el ADNMt(12). Sumado a esto, se describen permanentemente mutaciones nuevas en genes conocidos o en genes nuevos asociados a patología mitocondrial, por ello no es posible establecer con certeza el porcentaje de enfermedades causadas por el ADN nuclear o por el ADNMt. Las técnicas diagnósticas tradicionales pueden no ser suficientes y muchos pacientes pueden quedar sin diagnóstico(13). Por esto se desarrollan nuevas técnicas en las que se estudian paneles de genes por técnica de secuenciación de próxima generación, que permite confirmar el diagnóstico genético entre el 22% y 60% de los pacientes(12). Además, los haplotipos del ADNMt pueden modular los efectos patológicos de mutaciones en el ADN nuclear(14), pudiendo ser la misma mutación deletérea o beneficiosa, dependiendo del resto del genoma de base y del ambiente(15).
Riesgos reproductivos
Las enfermedades mitocondriales tienen complejas implicancias en la reproducción, ya que los riesgos de repetición en la familia dependerán de un estudio complejo del paciente, que determinará el patrón de herencia y la identificación de las mutaciones en el caso índice. En los casos de herencia mitocondrial, la posibilidad de recurrencia dependerá del estado de portadora de la madre. Algunas mutaciones ocurren de novo y la madre no es portadora; en ese caso se estima un riesgo de repetición alrededor de 1/24 (4%)(16). Si la madre es portadora de la mutación mitocondrial, habría un riesgo de transmisión variable. Habría varios factores que intervendrían en la variabilidad; por un lado la replicación selectiva del genoma y, por otro, el efecto de cuello de botella, que resulta en una variable transmisión del porcentaje de mitocondrias mutadas a la descendencia de las madres heteroplásmicas(17). Las madres heteroplásticas pueden tener óvulos con un porcentaje variable de mitocondrias mutadas y, a su vez, distribuirse en forma irregular en los distintos tejidos embrionarios.
Existen distintas estrategias para familias en riesgo de tener hijos con enfermedad mitocondrial en los aspectos reproductivos(18):
1. La dación de ovocitos con una dadora no portadora( 19). De esta manera se asegura que el niño nazca sin la patología mitocondrial, pero no tendría vínculo biológico con la madre en riesgo gestante.
2. La selección de embriones o fetos con menor riesgo de padecer la enfermedad. Es aplicable a las madres con heteroplasmía y cuando hay una buena correlación entre la tasa de mitocondrias mutadas y el fenotipo(20):

– El diagnóstico prenatal, mediante el estudio de la heteroplasmía en células de vellosidades coriales( 21). Este estudio puede informar si el riesgo es mayor o menor; sin embargo, dado que no se estudia el tejido fetal sino el trofoblasto, es un resultado orientador acerca del porcentaje de mitocondrias mutadas que pudo haber heredado el niño, pero no es posible determinar la distribución de dichas mitocondrias en los diferentes tejidos, debido a los fenómenos de cuello de botella y replicación selectiva de las mitocondrias.
– El diagnóstico de preimplantación tiene la finalidad de seleccionar un embrión con menor carga de mitocondrias mutadas. Luego de la fertilización in vitro, al tercer día, en el estado de mórula, se biopsia al embrión y se analiza la carga de mitocondrias mutadas, y debido a que la variación entre las blastómeras es baja, se puede estimar el riesgo de que el embrión desarrolle la enfermedad mitocondrial(22). Si se realiza la biopsia del trofoblasto, al quinto día, en estado de blastocisto, las células obtenidas pueden no reflejar totalmente el grado de heteroplasmía, dando lugar a resultados de baja confiabilidad(23).
En resumen, el diagnóstico preimplantatorio en la mórula sería más confiable, no así en blastocisto o el diagnóstico prenatal en células de vellosidades que generarían más incertezas(18), pudiendo ofrecerse estas técnicas sólo a pacientes con heteroplasmía.
Las limitaciones de estas técnicas diagnósticas y la falta de relación biológica en el caso de la ovodación llevaron a la búsqueda de nuevas estrategias para evitar el nacimiento de niños con patología mitocondrial.
Dos técnicas se han propuesto recientemente: la trasferencia pronuclear (ambos corpúsculos polares luego de la fecundación) y la transferencia del huso meiótico cromosómico (detenido en la metafase II, antes de la fecundación). Ambos procedimientos tienen como finalidad transferir el genoma nuclear del embrión o del ovocito a un ambiente mitocondrial funcional. En el primer caso, el genoma nuclear embrionario (ambos pronúcleos materno y paterno) se transfiere a otro embrión enucleado gestado con un ovocito de una mujer dadora sin enfermedad mitocondrial y el semen del padre; en el segundo, el genoma nuclear materno (huso meiótico cromosómico) se transfiere a un ovocito enucleado dado por una mujer sin enfermedad mitocondrial(22).

Estos procedimientos tienen como finalidad asegurar que el embrión reciba el genoma nuclear parental en un nuevo ambiente citoplasmástico, con mitocondrias funcionantes.
La transferencia pronuclear implica la remoción de un pequeño segmento de citoplasma con ambos pronúcleos del cigoto con las mitocondrias mutadas (carioplasto) y su transferencia al espacio perivitelino de un cigoto enucleado con mitocondrias normales. Esta transferencia requiere la fusión del carioplasto removido mediante pulsos eléctricos o a virus hemaglutinante del Japón (HVJ o virus Sendai). Estos estudios se han realizado en un modelo animal de ratón(24) y en embriones humanos anormales gestados por tratamientos de fertilidad que fueron dados para investigación. De estos embriones, solo el 8,3% desarrollaron a la etapa de blastocisto, con un porcentaje <2% de mitocondrias mutadas, pero con distribución desigual entre las blastómeras(25).
En el segundo caso, la transferencia del huso meiótico cromosómico se ha realizado en primates(26) y en embriones humanos que progresaron hasta estado de blastocisto, cuyas células portaban <1% de mitocondria mutadas(27).
Aspectos científicos
Por el momento, no se conoce la seguridad y eficacia de estas técnicas aplicadas a seres humanos. Existen muchos aspectos científicos a considerar, entre ellos:
Mitocondriales mutadas residuales:
El fenómeno de arrastre de mitocondrias mutadas junto con el genoma nuclear(28), descrito entre un 0,01-2% en distintas publicaciones, generando ello un grado variable de heteroplasmía( 18). Si bien este porcentaje no sería suficiente para generar una enfermedad(25), aún no hay evidencia de cómo se distribuirán estas mitocondrias en los tejidos fetales y cómo afectará la heteroplasmía el desarrollo del embrión o a su descendencia en caso de ser una niña(5).

Manipulación embrionaria:
Es necesario determinar si la manipulación del embrión asociada al transplante del genoma nuclear puede afectar el desarrollo embrionario normal. Por sí mismas, las técnicas de fertilización asistida incrementan el riesgo de anomalías congénitas, autismo y de trastorno epigenéticos(29-34).
Aún más, la manipulación del huso meiótico durante el transplante puede incrementar el riesgo de pérdida de cromosomas durante su manipulación, que afecta el desarrollo embrionario, ya que solo sería posible ver los cromosomas con el uso de una tinción fluorescente(35).
Todavía no hay datos específicos para estas técnicas sobre anomalías congénitas, cromosómicas o efectos de imprinting en los trabajos realizados. El trabajo de Craven se realizó en embriones fertilizados in vitro, considerados anormales y no aptos para ser transferidos a su madre en tratamiento de fertilidad. Solo el 50% de estos embriones llegaron al estadio de blastocisto(25). Por el momento, existe un trabajo en transferencia de huso meiótico cromosómico en ovocitos humanos que desarrollan hasta etapa de blastocisto(36).
Además, la transferencia nuclear requiere el uso de inhibidores reversibles del citoesqueleto celular: inhibidores de la polimerización de la actina y tubulina en la transferencia de los pronúcleos, y de la actina del citoesqueleto en la transferencia del huso meiótico. No se conocen aún los efectos de estos inhibidores en el desarrollo embrionario(35).
Compatibilidad entre el genoma nuclear y el genoma mitocondrial:
Existe una comunicación cruzada, interacciones mitonucleares, entre el genoma mitocondrial y el nuclear, ya que el genoma nuclear codifica proteínas que intervienen en la función, mantenimiento y replicación de las mitocondrias(37). Muchos de estos genes se ubican en los autosomas y muy pocos en el cromosoma X, de manera de facilitar la coadaptación de ambos genomas; sin embargo, es conocido que algunas enfermedades mitocondriales tendrían una expresión más severa en los hijos que en las hijas, existiendo un factor de protección en los cromosomas X(38).

Ovocitos dados:
Alrededor de una de cada 400 personas portan mutaciones patogénicas del ADN mitocondrial heredadas vía materna. Estas mutaciones pueden ser asintomáticas o presentar síntomas con los años, como trastornos neurológicos, hipoacusia, neuropatía, diabetes, cardiomiopatía(39), lo que implicaría la necesidad de realizar una detallada historia clínica personal y familiar, y estudios genéticos en la dadora aún sin antecedentes relevantes para descartar que también sea portadora de alguna mutación del genoma mitocondrial.
A su vez, es necesario que haya una sincronización de la estimulación ovárica de la dadora y la paciente, de manera de obtener los óvulos el mismo día. Dadas las diferencias personales en la respuesta a la hiperestimulación ovárica, no siempre es posible esta sincronización y sería necesario congelar los ovocitos o los embriones. La vitrificación puede dañar el huso meiótico, por ello no es recomendable vitrificar el óvulo de la dadora para estos procedimientos(40).
Aspectos éticos
Estas dos técnicas, la trasferencia pronuclear y la transferencia del huso meiótico cromosómico diseñadas para la llamada “donación mitocondrial”, requieren la fertilización in vitro de embriones y la dación de óvulos por una tercera persona.
La transferencia del huso meiótico cromosómico implica la enucleación de sendos óvulos y la transferencia del material nuclear de la mujer portadora de la enfermedad mitocondrial al óvulo de la dadora, y la fertilización in vitro con el semen de la pareja de la mujer en riesgo.
En la trasferencia pronuclear, es necesario fecundar dos embriones: uno con el óvulo de la mujer en riesgo y otro embrión con el óvulo de la dadora. Estos dos seres humanos en etapa embrionaria serán privados de su desarrollo natural, ya que si se permitiera su transferencia al útero materno para continuar su desarrollo a término, en el primer caso podría nacer un niño con patología mitocondrial y, en el segundo, un niño sano. Pero en esta técnica se destruyen estos dos seres humanos en etapa embrionaria, con el objetivo de generar un tercer ser humano con el material genético y celular de ambos.

Existen muchos aspectos éticos a considerar, entre ellos:
La investigación en seres humanos en etapa embrionaria:
La definición de quiénes son los seres humanos que requieren mayor protección en este tipo experimental de técnicas de fertilización asistida: si son los padres, quienes sufren la profunda angustia de tener hijos con riesgo elevado de patología hereditaria, o son los mismos embriones, sus propios hijos, quienes, sometidos a esta necesidad de ser “sanos”, son expuestos a técnicas de investigación de las que aún no se conocen sus consecuencias y que implican un riesgo mayor que el riesgo mínimo(41). La pregunta es si la necesidad de tener un hijo relacionado biológicamente es tal que justifica, por un lado, concebir a sus propios hijos mediante una técnica experimental de la cual aún hay muy poca información; por otro lado, exponer a una tercera persona a los riesgos, si bien bajos, no inexistentes, de una estimulación ovárica; y finalmente someter a otros seres humanos en etapa embrionaria, no aptos o no queridos para su transferencia, a procesos de investigación que permitan validar la eficacia y seguridad de estas técnicas.
Gestación deliberada de embriones humanos como material biotecnológico:
En la técnica de la trasferencia pronuclear es necesario gestar dos embriones humanos que serán destruidos en forma deliberada, con el fin de obtener productos biotecnológicos (carioplasma y citoplasma) que serán utilizados para la producción de un tercer embrión. Se interpretan ambos seres humanos como un producto que será reacondicionado para poder ser considerado como un embrión “permitido” y así ser transferido a un útero materno(42); interpretando al embrión humano gestado con el óvulo de la dadora como un ser humano de descarte, cuya vida está destinada a ser sacrificada para el nacimiento de otro niño sano, con el consentimiento de sus progenitores. Este embrión se asume, en estas técnicas, como un producto biotecnológico de materia prima, perdiendo toda su identidad y dignidad humana. Ya no es un fin es sí mismo, es un medio que permitirá que otro niño —quien aún puede ser su medio hermano vía paterna— nazca sano.
A esto se agrega la selección de seres humanos en etapa embrionaria según sus características genéticas. Es una práctica deshumanizante, ya que se asientan en una mentalidad eugenésica. Por un lado, se descartan a los seres humanos con características genéticas no deseadas y, por otro, se mejoran las características genéticas de las personas, perdiendo su identidad. Si bien se modifica un porcentaje pequeño de la información genética, existe una importante conexión entre el ADN nuclear y el ADN mitocondrial que no solo se interviene en la función bioenérgetica de las mitocondrias en el funcionamiento celular, sino que se describe más frecuentemente la contribución mitocondrial a la capacidad de neuroplasticidad y al desarrollo de la personalidad(43,44).
Conclusiones
Estas modificaciones, que pretenden ser terapéuticas, no son inocuas ni están exentas de consecuencias desconocidas, y como tales deben interpretarse en una perspectiva más amplia, ya que estas técnicas no solo no alientan la investigación en el tratamiento de las personas con enfermedades mitocondriales, sino que evitan su nacimiento. El avance de la ciencia debe interpretarse como un medio que busque el bienestar de todas las personas, centrado en la protección de aquellos individuos más vulnerables de nuestra gran familia humana, en este caso los seres humanos en etapa embrionaria.
Referencias Bibliográficas
1. Koopman WJ, Willems PH, Smeitink JA. Monogenic mitochondrial disorders. N Engl J Med 2012; 366: 1132-141.
2. Vafai SB, Mootha VK. Mitochondrial disorders as windows into an ancient organelle. Nature 2012; 491: 374-383-
3. Taylor RW, Turnbull DM. Mitochondrial DNA mutations in human disease. Nat Rev Genet 2005; 6: 389- 402.
4. Greaves LC, Reeve AK, Taylor RW, Turnbull DM. Mitochondrial DNA and disease. J Pathol 2012; 226: 274-286.
5. Amato P, Tachibana M, Sparman M, Mitalipov S. Three-parent in vitro fertilization: gene replacement for the prevention of inherited mitochondrial diseases. Fertil Steril 2014; 101(1): 31-35.
6. Jeppesen TD, Schwartz M, Frederiksen AL,Wibrand F, Olsen DB, Vissing J. Muscle phenotype and mutation load in 51 persons with the 3243A>G mitochondrial DNA mutation. Arch Neurol 2006; 63: 1701-1706.
7. Chinnery PF. Mitochondrial Disorders Overview. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews ®. Seattle (WA): University of Washington, Seattle; 2015. Available from: http://www.ncbi.nlm. nih.gov/books/NBK1224/
8. Tuppen HA, Blakely EL, Turnbull DM, Taylor RW. Mitochondrial DNA mutations and human disease. Biochim Biophys Acta 2010; 1797: 113-128.
9. Werner JH, Koopman WJH, Willems PHGM, Smeitink JAM. Monogenic Mitochondrial Disorders. N Engl J Med 2012; 366: 1132-1141.
10. Arpa J, Cruz-Martínez A, Campos Y, Gutiérrez- Molina M, García-Rio F, Pérez-Conde C, Martín MA, Rubio JC, Del Hoyo P, Arpa-Fernández A, Arenas J. Prevalence and progression of mitochondrial diseases: a study of 50 patients. Muscle Nerve 2003; 28(6): 690-695.
11. Gorman GS, Schaefer AM, Ng Y, et al. Prevalence of nuclear and mtDNA mutations related to adult mitochondrial disease. Ann Neurol 2015;(4). doi:10.1002/ana.24362.
12. Lieber DS, Calvo SE, Shanahan K, etal. Targeted exome sequencing of suspected mitochondrial disorders. Neurology 2013; 80:1762-1770.
13. Finsterer J, Jarius C, Eichberger H. Phenotype variability in 130 adult patients with respiratory chain disorders. J Inherit Metab Dis 2001; 24:560-576.
14. Strauss KA, DuBiner L, Simon M, et al. Severity of cardiomyopathy associated with adenine nucleotide translocator-1 deficiency correlates with mtDNA haplogroup. Proc Natl Acad Sci USA 2013; 110(9): 3453-3458
15. Ji F, Sharpley MS, Derbeneva O, et al. Mitochondrial DNA variant associated with Leber hereditary optic neuropathy and high-altitude Tibetans. Proc Natl Acad Sci USA 2012; 109: 7391-7396.
16. Chinnery PF, DiMauro S, Shanske S, et al. Risk of developing a mitochondrial DNA deletion disorder. Lancet 2004; 364: 592-596.
17. Bitner-Glindzicz M, Pembrey M, Duncan A, et al. Prevalence of mitochondrial 1555A–>G mutation in European children. N Engl J Med 2009; 360: 640-642.
18. Burgstaller JP, Johnston IG, Poulton J. Mitochondrial DNA disease and developmental implications for reproductive strategies. Mol Hum Reprod 015; 21(1): 11-22.
19. Sauer MV, Kavic SM. Oocyte and embryo donation 2006: reviewing two decades of innovation and controversy. Reprod Biomed Online 2006; 12: 153-162.
20. White SL, Collins VR, Wolfe R, et al. Genetic counseling and prenatal diagnosis for the mitochondrial DNA mutations at nucleotide 8993. Am J Hum Genet 1999; 65: 474-482.
21. Harding AE, Holt IJ, Sweeney MG, Brockington M, Davis MB. Prenatal diagnosis of mitochondrial DNA8993 T—G disease. Am J Hum Genet 1992; 50: 629-633.
22. Poulton J, Oakeshott P. Nuclear transfer to prevent maternal transmission of mitochondrial DNA disease. BMJ 2012; 345: e6651.
23. Treff NR, Campos J, Tao X, Levy B, Ferry KM, Scott RT Jr. Blastocyst preimplantation genetic diagnosis (PGD) of a mitochondrial DNA disorder. Fertil Steril 2012; 98: 1236-1240.
24. McGrath J, Solter D. Nuclear transplantation in the mouse embryo by microsurgery and cell fusion. Science 1983; 220: 1300-1302.
25. Craven L, Tuppen HA, Greggains GD, et al. Pronuclear transfer in human embryos to prevent transmission of mitochondrial DNA disease. Nature 2010; 465: 82-85.
26. Tachibana M, Sparman M, Sritanaudomchai H, et al. Mitochondrial gene replacement in primate offspring and embryonic stem cells. Nature 2009; 461: 367-372.
27. Tachibana M, Amato P, Sparman M, et al. Towards germline gene therapy of inherited mitochondrial diseases. Nature 2013; 493: 627-631.
28. St John JC, Campbell KH. The battle to prevent the transmission of mitochondrial DNA disease: is karyoplast transfer the answer? Gene Ther 2010; 17: 147-149.
29. Davies MJ, Moore VM, Willson KJ, et al. Reproductive technologies and the risk of birth defects. N Engl J Med 2012; 366(19): 1803-1813.
30. Jafarzadehpur E, Kermani RM, Mohhamadi AR, et al. Family Ocular Manifestations in Infants Resulted from Assisted Reproductive Technology (ART). Reprod Health 2013; 7(4): 181-186.
31. Tarín JJ, García-Pérez MA, Cano A. Assisted reproductive technology results: why are live-birth percentages so low? Mol Reprod Dev 2014; 81(7): 568-583.
32. Uyar A, Seli E. The impact of assisted reproductive technologies on genomic imprinting and imprinting disorders. Curr Opin Obstet Gynecol 2014; 26(3): 210-221.
33. Gutarra-Vilchez R, Santamariña-Rubio E, Salvador J, Borrell A. Birth defects in medically assisted reproduction pregnancies in the city of Barcelona. Prenat Diagn 2014; 34(4): 327-334.
34. Kissin DM, Zhang Y, Boulet SL, et al. Association of assisted reproductive technology (ART) treatment and parental infertility diagnosis with autism in ART-conceived children. Hum Reprod 2015; 30(2): 454-465
35. Craven L, Elson JL, Irving L, et al. Mitochondrial DNA disease: new options for prevention. Hum Mol Genet 2011; 20(2): 168-174.
36. Tanaka A, Nagayoshi M, Awata S, et al. Metaphase II karyoplast transfer from human in-vitro matured oocytes to enucleated mature oocytes. Reprod Biomed Online 2009; 19: 514-520.
37. Wolff JN, Ladoukakis ED, Enríquez JA, Dowling DK. Mitonuclear interactions: evolutionary consequences over multiple biological scales. Philos Trans R Soc Lond B Biol Sci 2014; 369(1646): DOI: 10.1098/ rstb.2013.0443.
38. Rogell B, Dean R, Lemos B, Dowling DK. Mito-nuclear interactions as drivers of gene movement on and off the X-chromosome. BMC Genomics 2014; 2(15): 330.
39. Poulton J, Oakeshott P. Nuclear transfer to prevent maternal transmission of mitochondrial DNA disease. BMJ 2012; 345: 6651.
40. Zarei Moradi S, Mohseni Meybodi A, et al. Chromosome Abnormalities and Viability of Vitrified Eight- Cell Mouse Embryos at Presence of Two Different Cryoprotectants at Different Storage Durations. Cell Journal (Yakhteh) 2013; 14(4): 254-263.
41. Strong C. Minimal Risk in Research Involving Pregnant Women and Fetuses. J Law Med Ethics 2011; 39(3): 529-538.
42. Health Science and Bioethics Division, Department of Health. Mitochondrial Donation: Government response to the consultation on draft regulations to permit the use of new treatment techniques to prevent the transmission of a serious mitochondrial disease from mother to child. Disponible en: https://www.gov.uk/government/uploads/system/uploads/attachment_data/file/332881/Consultation_response.pdf
43. Klinedinst NJ, Regenold WT. A mitochondrial bioenergetic basis of depression. J Bioenerg Biomembr 2015; 47(1-2): 155-171.
44. Streck EL, Gonçalves CL, Furlanetto CB, et al. Mitochondria and the central nervous system: searching for a pathophysiological basis of psychiatric disorders. Rev Bras Psiquiatr 2014; 36(2): 156-167.
Más notas de la edición 66

Lee desde Issuu nuestra última edición publicada en Julio 2024, Edición número 155

Notas relacionadas a Valoración ética de las nuevas opciones...

