
Mucopolisacaridosis: características clínicas, diagnóstico y de manejo
Jorge Luis Suarez-Guerreroa,* , Pedro José Iván Gómez Higuerab, Juan Sebastian Arias Flórezb, Gustavo Adolfo Contreras-Garcíac
a UIS-HUS, Grupo de investigación en Genética Humana, Facultad de Salud, Universidad Industrial de Santander, Bucaramanga, Colombia
b Facultad de Salud, Universidad Industrial de Santander, Bucaramanga, Colombia
c Grupo de investigación en Genética Humana, Universidad Industrial de Santander, Departamento de Pediatría-Hospital Universitario de Santander, Bucaramanga, Colombia
Correspondencia a: Correspondencia a: Jorge Luis Suarez-Guerrero
Correo electrónico: jorgesuarezg_@hotmail.com, jorgesuarezg@gmail.com, jsuarezguerrero@yahoo.es
0370-4106/© 2015 Publicado por Elsevier España, S.L.U. en nombre de Sociedad Chilena de Pediatría.
Este es un artículo Open Access bajo la licencia CC BY-NC-ND
(http://creativecommons.org/licenses/by-nc-nd/4.0/).
Rev. chil. pediatr. vol.87 no.4 Santiago ago. 2016
http://dx.doi.org/10.1016/j.rchipe.2015.10.004
Alcalde Eduardo Castillo Velasco 1838
Ñuñoa, Santiago
Casilla 593-11
Tel.: (56-2) 2237 1598 -2237 9757
Fax: (56-2) 2238 0046
revista@sochipe.cl
Resumen
Las mucopolisacaridosis (MPS) son un grupo de enfermedades raras (huérfanas), de baja prevalencia, caracterizadas por la deficiencia de enzimas que participan en el metabolismo de glucosaminglucanos (GAG) a nivel lisosomal. Se caracteriza por acumulación de GAG intracelular, produciendo alteraciones de múltiples órganos y sistemas. Su diagnóstico se basa en el conocimiento de las manifestaciones clínicas, realizar el análisis bioquímico para identificar el tipo de GAG que se está acumulando y confirmar el tipo de enfermedad con la determinación enzimática correspondiente. Su identificación es fundamental para iniciar un tratamiento oportuno, teniendo en cuenta que actualmente existe manejo transdisciplinario y tratamiento de reemplazo enzimático para MPS I (síndrome de Hurler), MPS II (síndrome de Hunter), MPS IV (síndrome de Morquio) y MPS VI (síndrome de Maroteaux-Lamy). En esta revisión se analizan cada uno de estos síndromes, su diagnóstico y tratamiento.
Palabras clave: Mucopolisacaridosis. Síndrome de Sanfilippo. Síndrome de Hurler. Síndrome de Hurler-Scheie. Síndrome de Hunter. Síndrome de Sly.
Abstract
The mucopolysaccharidoses (MPS) are a group of rare (orphan) diseases, characterised by a deficiency of enzymes involved in the metabolism of glycosaminoglycans (GAGs) at lysosomal level. When there is a deficiency of a particular enzyme there is an accumulation of GAGs in the cells resulting in progressive cellular damage, which can affect multiple organ systems and lead to organ failure. Diagnosis is based on knowledge of the clinical manifestations, performing biochemical analyses to identify the type of GAG that is accumulating, and confirm the type of disorder with the corresponding enzymatic determination. Their identification is essential to initiate early treatment, taking into account that multidisciplinary management and enzyme replacement therapy is available for MPS I (Hurler syndrome), MPS II (Hunter syndrome), MPS IV (Morquio syndrome), and MPS VI (Maroteaux-Lamy syndrome. In this review, an analysis is made of each of these syndromes, as well as their diagnosis and treatment.
Keywords: Mucopolysaccharidosis. Sanfilippo syndrome. Hurler syndrome. Hurler-Scheie syndrome. Hunter syndrome. Sly syndrome.
Introducción
Las mucopolisacaridosis (MPS) pertenecen a un grupo heterogéneo de enfermedades que se generan por deficiencias enzimáticas, caracterizadas por la acumulación lisosomal de sustancias intermedias del metabolismo de los mucopolisacáridos o glucosaminoglucanos (GAG), que son macromoléculas que proporcionan soporte estructural a la matriz extracelular y son parte importante de los procesos de regulación y comunicación celular1,2. Estas enfermedades pertenecen al subgrupo de las patologías monogénicas, que en algunos casos pueden darse también por rearreglos cromosómicos.
El primer autor que identificó esta condición fue John Thomson en Edinburgo en 19083; sin embargo, los primeros casos publicados de MPS fueron en 1917 por Charles Hunter4. En 1919 publica Gertrud Hurler5 y en 1929 Luis Morquio reportó los primeros casos de la que hoy conocemos como MPS IV6. En 1936 Ellis, Sheldon y Capon indican el término «gargolismo», al comparar las características craneales, faciales y torácicas con la figuras llamadas «gárgolas» de la Catedral de Notre Dame en París7. Solo hasta 1952 Brante aisló las primeras muestras de MPS en 2 pacientes con síndrome de Hunter, y para 1970 se aislaron el dermatán y heparán sulfato.
La clínica, edad de presentación, diagnóstico, tratamiento y complicaciones varían entre una MPS a otra e incluso dentro del espectro de una misma MPS, requiriendo trabajo transdisciplinar para diagnosticar, tratar y orientar a sus familias.
El objetivo de esta revisión es actualizar el conocimiento sobre las manifestaciones clínicas, diagnóstico y manejo de las diferentes formas de esta patología. Como método de búsqueda se llevó a cabo una investigación sistemática en las bases de datos PubMed y Scielo, usando los términos MPS, Mucopolysaccharidosis (PubMed) o mucopolisacaridosis (scielo) con su respectiva numeración y por su epónimo.
Enfermedades lisosomales
Son un grupo de aproximadamente 50 procesos patológicos diferentes con una prevalencia general que varía entre 1/1.500 a 1/7.000 nacidos vivos (sumando todas las enfermedades del grupo). Desde el punto de vista epidemiológico afecta a todos los grupos étnicos género por igual, excepto en los casos en que la condición está ligada al cromosoma X. Principalmente se aprecian en la infancia, pero varias formas atenuadas se diagnostican en adultos. Las manifestaciones clínicas dependerán del defecto enzimático y la expresión diferencial en órganos y sistemas8,9.
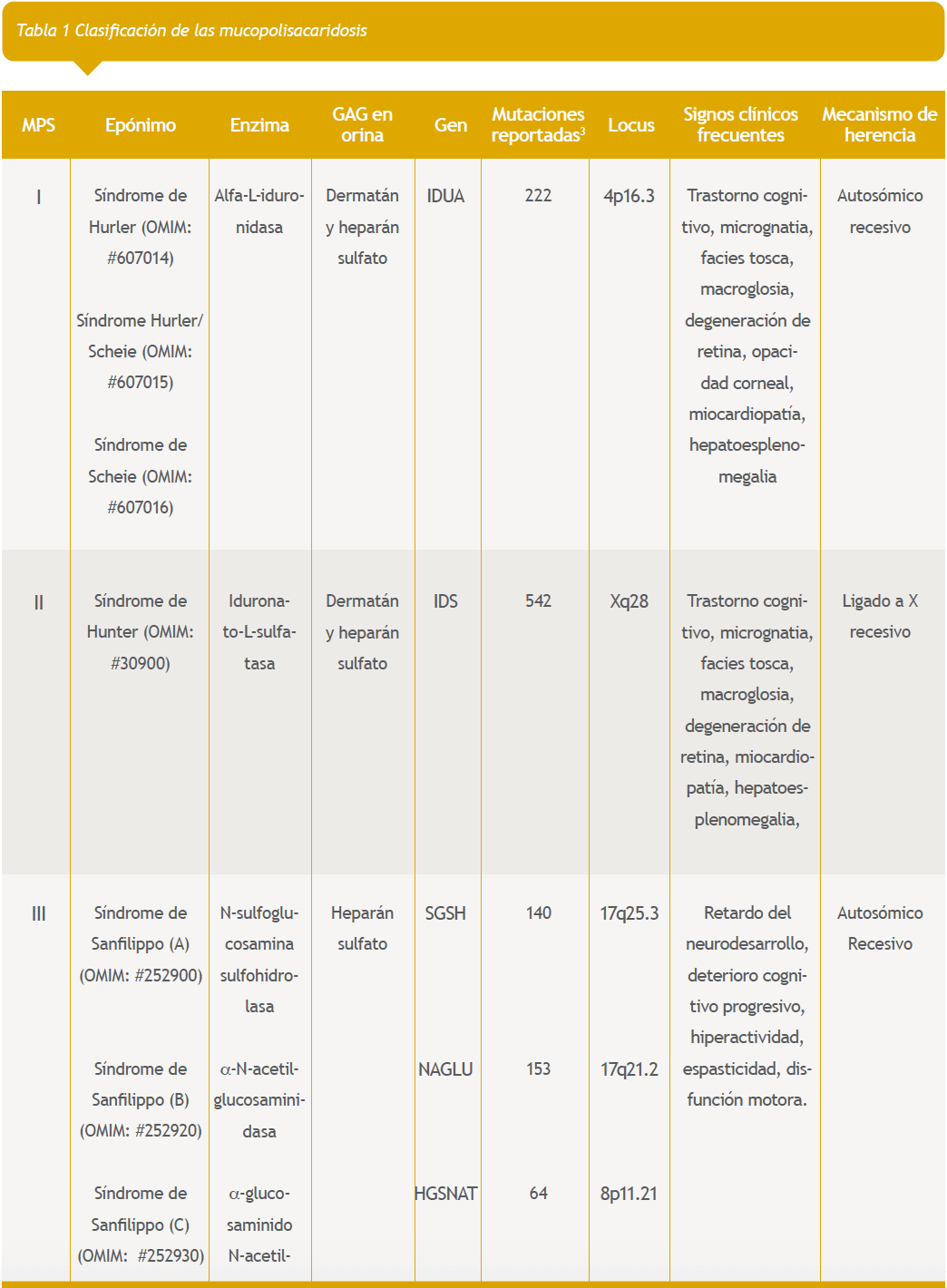
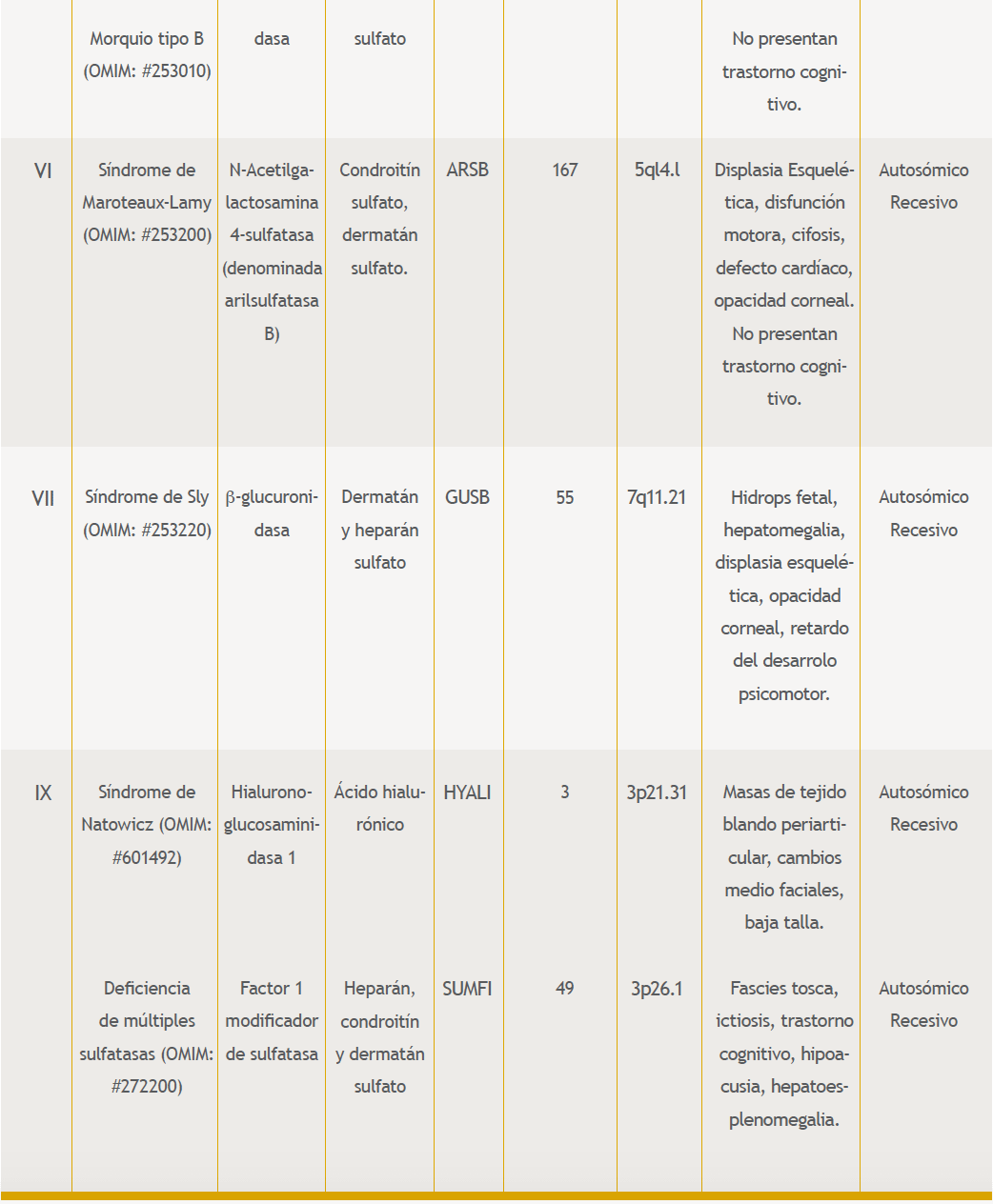
En este grupo se encuentra el subgrupo de las MPS, las cuales son MPS I o síndrome de Hurler (Hurler, Hurler-Scheie y Scheie), MPS II o síndrome de Hunter (variantes A y B), MPS III o síndrome de Sanfilipo (variantes A-D), MPS IV o síndrome de Morquio (variantes A y B), MPS VI o Marotaux-Lamy, MPS VII o síndrome de Sly, MPS IX o síndrome de Natowicz2,10 y las deficiencias de múltiples sulfatasas (ver todas en la tabla 1). Los datos epidemiológicos para MPS se desconocen en nuestro país, pero en Europa y Australia se tienen algunos estimados, así por ejemplo en Europa la ocurrencia es de 1:10.000 a 1:25.000 nacidos vivos.

Mucopolisacaridosis I (Hurler, Hurler-Scheie y Scheie)
En la MPS I hay acumulación de heparán sulfato (HS) y dermatán sulfato (DS), lo cual genera disfunción multiorgánica progresiva11–16. La enfermedad se ha clasificado en 3 fenotipos:

- MPS IH o síndrome de Hurler, caracterizado por síntomas severos tempranos que generan el deceso en la primera década de vida.
- MPS IH/S o síndrome de Hurler-Scheie, presenta menores complicaciones que el fenotipo MPS IH y las personas son más longevas, falleciendo entre la segunda o tercera década de vida.
- MPS IS o síndrome de Scheie (antes MPS tipo V, luego se descubrió que el defecto enzimático era el mismo que Hurler)11 se presenta con síntomas atenuados y buen pronóstico de vida, viviendo alrededor de los 30 a 40 años.
Manifestaciones clínicas
Existe afectación multiorgánica, las principales características son: baja talla, facies tosca, macrocefalia, macroglosia, labios gruesos, cejas pobladas, opacidad corneal, glaucoma, hidrocefalia comunicante, compresión medular, síndrome del túnel carpiano, pérdida de la audición, hepatoesplenomegalia, obstrucción de las vías aéreas con la consecuente apnea o disnea, enfermedades cardiacas, hernias umbilicales e inguinales, rigidez articular y disostosis múltiple2,5,14–17. Respecto al neurodesarrollo, la MPS IH es la única que produce trastorno cognitivo, en las otras 2 variantes existe un desarrollo cognitivo cercano a lo normal17. Los casos severos son diagnosticados con relativa facilidad por el fenotipo tan llamativo, sin embargo las formas atenuadas que generan Scheie son más difíciles de identificar, ya que pueden presentarse solo como enfermedad articular que en ocasiones se confunde con enfermedades reumatológicas de la infancia18.
Complicaciones
El aumento de depósito de DS y HS en órganos favorece la aparición paulatina de complicaciones, entre ellas opacidad corneal con posterior pérdida de la visión, enfermedad pulmonar restrictiva, rinitis crónica recurrente acompañada de secreción nasal persistente e infecciones frecuentes del oído, hipoacusia conductiva y neurosensorial, hipertensión endocraneal. Cardiopatías como el engrosamiento de la válvula cardíaca, estenosis aórtica, insuficiencia cardíaca y arritmias. La muerte ocurre por enfermedad obstructiva de la vía aérea, infecciones respiratorias o complicaciones cardíacas.
Mucopolisacaridosis II (Hunter)
Afecta con mayor proporción a hombres que a mujeres, por presentar un patrón de herencia recesivo ligado al cromosoma X14,19,20. La enzima iduronato-L-sulfatasa mutada genera alteraciones en procesos de clivaje de las fracciones sulfatadas del DS y del heparán sulfato (HS).

Manifestaciones clínicas
La MPS II se subdivide en 2 grandes categorías, la MPS IIA (severa) y la MPS IIB (moderada). La aparición de las manifestaciones clínicas en la MPS IIA es en los primeros meses, mientras que en MPS IIB de los 3-4 años. Las principales características son facies tosca, opacidades corneales, macroglosia, rinorrea, hipertrofia gingival, hirsutismo, cuello y tórax cortos, manos en garra, hipercifosis lumbar, hernias inguinal y umbilicales, hepatoesplenomegalia leve, engrosamiento de tejidos blandos y cartílagos, con la consecuente disostosis ósea, apneas del sueño, valvulopatías y miocardiopatías19–21. La afección del sistema nervioso varía, en la tipo IIA se presenta trastorno cognitivo progresivo, hiperactividad y agresividad, mientras que en la tipo IIB hay desarrollo neuronal normal20.
Complicaciones
Los depósitos orofaríngeos obstruyen la cavidad supraglótica favoreciendo la apnea del sueño, y los traqueobronquiales facilitan infecciones en el tracto respiratorio; pueden presentar otitis media, hipoacusia, mielopatia cervical y compresión medular19–21. Respecto a las anomalías esqueléticas es común encontrar artropatía de cadera, articulaciones rígidas, síndrome de túnel carpiano, mano en garra, algunos requiriendo corrección quirúrgica19,20. La mortalidad en casos graves suele presentarse en la segunda década de vida secundaria a fallas cardiacas y respiratorias14,19,20, mientras que los leves son más longevos.
Mucopolisacaridosis III (Sanfilippo)
Existen 4 subtipos de MPS III, descritos en la tabla 1 con sus respectivas enzimas lisosomales. Los signos no varían mucho, y a pesar de los intentos de separar los rasgos fenotípicos de cada subtipo, no ha sido posible debido a la inmensa heterogeneidad alélica y polimorfismos. Las MPS III más comunes son los subtipos A y B, siendo el C y D subtipos pocos frecuentes en la clínica22.

Manifestaciones clínicas
Las manifestaciones clínicas, aunque son muy semejantes, pueden variar entre fenotipos23. El retraso neurológico y la degeneración del sistema nervioso central son evidentes a partir de los 6 a 10 años de edad, con retardo del lenguaje, hiperactividad, agresividad y trastornos del sueño22. Los rasgos dismorfológicos no son tan evidentes, presentando así facies toscas pero de manera sutil, macroglosia, cejas pobladas, labio inferior grueso y evertido, dolicocefalia, surco nasolabial prominente, hirsutismo, hipoacusia, otitis e infecciones en la garganta22,24,25. Otros síntomas menos frecuentes son hepatomegalia, macrocefalia o las hernias inguinales. El crecimiento puede ser normal, las anormalidades cardiacas o esqueléticas son raras.
Complicaciones
Las complicaciones se producen en 3 fases. La primera fase comienza antes de los 3 años, la cual se caracteriza por hipoacusia, retraso en el lenguaje, falta de control de esfínteres, otitis, faringitis y diarrea22. La segunda fase se inicia entre los 3-4 años y se hacen evidentes los trastornos en el sueño, con hiperactividad y agresividad; en pocos casos aparecen signos como escoliosis, cifosis, lordosis lumbar y síndrome del túnel carpiano22. La tercera fase aparece hacia la primera década de vida, donde la hiperactividad cesa, aumenta la espasticidad, pérdida de equilibrio, convulsiones, hasta la pérdida importante de movilidad que genera incapacidad permanente; surgen problemas para deglutir y alimenticios, por lo que su ingesta es líquida y en algunos casos con sonda nasogástrica22. Los pacientes fallecen entre la segunda y la tercera décadas de la vida, usualmente por infecciones respiratorias severas22,24,26.
Mucopolisacaridosis tipo IV (Morquio)
Existen 2 subtipos de MPS IV, la MPS IVA con una frecuencia variable a nivel mundial de 1/75.000 a 1/200.0001,27. Existe afección del sistema osteoarticular y del tejido de sostén por alteración del metabolismo del queratán sulfato y condroitín sulfato, generando daño permanente y progresivo subsecuente en fibroblastos y leucocitos. El gen afectado GALNS codifica para la enzima GALNS encargada de hidrolizar el condroitín sulfato de la N-acetil-D-galactosamina 6-sulfato y las unidades de queratán sulfato de la D-galactosa 6-sulfato28–30.

Por su parte, la MPS IVB, cuyo resultado es la acumulación de queratán sulfato, presenta manifestaciones clínicas similares a la MPS IV A31. Su prevalencia estimada varía de 1/75.000 a 1/640.000 nacidos vivos31.
Manifestaciones clínicas
Las alteraciones aparecen entre el primer a tercer año de vida1, con baja talla, tronco corto, pectus carinatum, cifoescoliosis, hiperlaxitud, inestabilidad de la columna cervical y vértebras en cuña u ovoides1,2,27,32–34; a nivel craneofacial presentan facies tosca (no muy marcada), prognatismo, boca amplia, puente nasal plano, opacidades en la córnea, malformaciones y caries en los dientes, hipoacusia, cuello corto e hipoplasia odontoidea1,27,34. Se presenta hiperlaxitud articular a nivel de cadera y en extremidades inferiores genu valgo y pie plano33–35. La radiografía muestra metacarpos cortos y anchos, coxa valga, cabezas femorales pequeñas o aplanadas27. El desarrollo psicomotor y cociente intelectual se conserva27,32,33,35,36; sin embargo, las alteraciones vertebrales pueden comprimir la médula generando debilidad progresiva y parálisis2,27,34.
Complicaciones
Varían dependiendo de la severidad de la enfermedad y de la edad del paciente; sin embargo, en general se pueden citar: plejías secundarias a lesiones cervicales por hipoplasia odontoidea, restricciones respiratorias por las malformaciones de tórax, escoliosis y hepato o esplenomegalia.
Mucopolisacaridosis tipo VI (Maroteaux-Lamy)
La mucopolisacaridosis tipo VI genera acumulación de DS. Se estima una prevalencia que puede variar entre 1 en 43.261 hasta 1 en 1.505.160 nacidos vivos14,37–40.
Características clínicas
Debido a la acumulación de DS se presentan 2 espectros de la enfermedad, la leve o lenta y la severa o rápida38, cada una con diversas complicaciones. La severa comienza generalmente antes de los 2 años de edad, presentando complicaciones cardiacas38,41 que llevan al deceso entre la segunda o tercera décadas de la vida. La forma leve comienza tardíamente, siendo las alteraciones músculo-esqueléticas leves las primeras en aparecer clínicamente y falleciendo entre la cuarta o quinta década de vida. Entre las principales características clínicas para ambos espectros se encuentran: baja talla para la edad, disostosis múltiple (como principal característica de la enfermedad en su forma severa)38, rigidez articular, opacidad corneal, facies tosca14,38 y a nivel cardiovascular valvulopatías y miocardiopatías como principal causa de muerte14,38,41.
De forma poco frecuente se pueden presentar macrocefalia, frente prominente, puente nasal deprimido, compromiso pulmonar14,38,41 y hernias inguinales o umbilicales. El desarrollo cognitivo usualmente es normal14,38.
Complicaciones
La acumulación del DS lleva a varias complicaciones a nivel pulmonar extrínsecas e intrínsecas, hepatomegalia, opacidad corneal, glaucoma y el edema de papila con atrofia óptica se aprecia en MPS VI avanzada. Alteraciones cardiacas como las estenosis y/o las insuficiencias en las válvulas mitral, tricúspide y aórtica son muy características de esta MPS; además, otras como la endocarditis y la hipertrofia ventricular pueden estar presentes en estas personas38,41.
Mucopolisacaridosis tipo VII (Sly)
La mucopolisacaridosis tipo VII está caracterizada por la acumulación de ácido glucorónico, debido a la deficiencia de la beta-glucoronidasa, con prevalencia de 1 en 250.000 nacidos vivos.
Características clínicas
Las características principales son la baja talla, macrocefalia, facies tosca, cuello corto, opacidades corneales, pectus carinatum, cifosis, escoliosis, hepatomegalia, esplenomegalia, hernia umbilical, hernia inguinal, disostosis múltiple, hipoplasia odontoidea, hipertricosis, displasia acetabular, articulaciones contraídas, retardo del neurodesarrollo, hidrocefalia y como manifestación prenatal se encuentra el hidrops fetal42,43.
Complicaciones
El fenotipo más severo presenta hidrops fetal, y su expectativa de vida es de meses, mientras que su forma leve puede llegar a la quinta década de la vida. Esta MPS comparte complicaciones como las hernias inguinales y umbilicales, la hepatomegalia, la esplenomegalia y la opacidad corneal. Además, existen frecuentes infecciones respiratorias y retardo del neurodesarrollo marcado en su forma grave43-45.
Mucopolisacaridosis tipo IX (Natowicz)
En la mucopolisacaridosis tipo IX existe depósito lisosomal de ácido hialurónico.
Características clínicas
Se caracteriza por baja talla, úvula bífida, paladar hendido, puente nasal deprimido, acumulación de masas en tejido blando periarticular, cambios medio faciales, otitis media y un signo casi patognomónico, que son las erosiones acetabulares18.
Complicaciones
Por la baja frecuencia de esta enfermedad no se reportan complicaciones diferentes a las manifestaciones clínicas. Se pueden prever complicaciones relacionadas con las altas concentraciones de ácido hialurónico en el líquido sinovial y en los tejidos sólidos, como en el cartílago y la piel. Estas altas concentraciones en los tejidos generarán directamente las manifestaciones clínicas10.
Deficiencia de múltiples sulfatasas
La deficiencia de múltiples sulfatasas o mucosulfatidosis se caracteriza por el depósito de esfingolípidos, esteroides y glucosaminoglucanos sulfatados.
Características clínicas
Las personas afectadas pueden presentar baja talla, facies tosca, frente amplia, aplanamiento facial, pérdida de la audición, disostosis múltiple, hipotonía neonatal, ictiosis severa, hepatomegalia, esplenomegalia, con importante retardo del desarrollo psicomotor y mental, ataxia e hiperreflexia de miembros inferiores. Según la edad de aparición y severidad de la enfermedad se ha clasificado en las variantes neonatal-infancia tardía (0-2 años) y juvenil (2-4 años)46–49.
Complicaciones
Las personas con la variante neonatal-infancia tardía (0-2 años) suelen presentar mayores complicaciones y fallecimiento en la infancia.
Diagnóstico de las mucopolisacaridosis
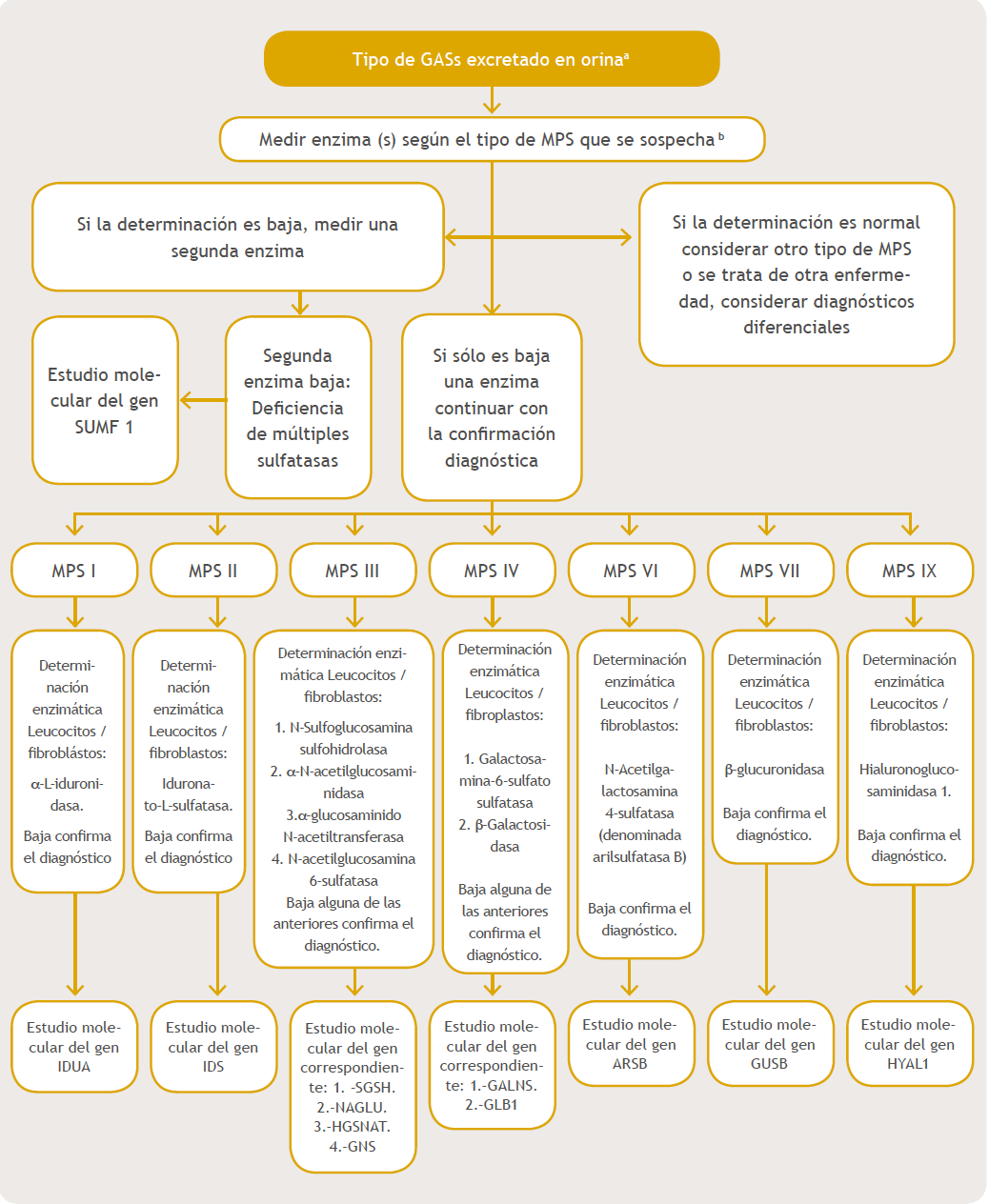
El diagnóstico de estas enfermedades se inicia con una historia clínica completa, especificando en antecedentes, si existen otros miembros en la familia con las mismas características clínicas, y si existe consanguinidad parental, por tener mecanismos de herencia autosómico recesivo, con excepción de MPS II que es ligada a X recesiva. La edad de inicio de las manifestaciones clínicas y un orden cronológico de la evolución, así como de las complicaciones, son datos fundamentales para poder orientar el diagnóstico. Los signos clínicos son el punto de partida para la orientación diagnóstica y aproximación al tipo de MPS que presenta el paciente19–21. Los hallazgos de imágenes diagnósticas complementan el estudio inicial, siendo los principales disostosis ósea evaluada por radiología y anormalidad en la sustancia blanca y espacios perivasculares en los casos graves evaluados por resonancia magnética cerebral20.
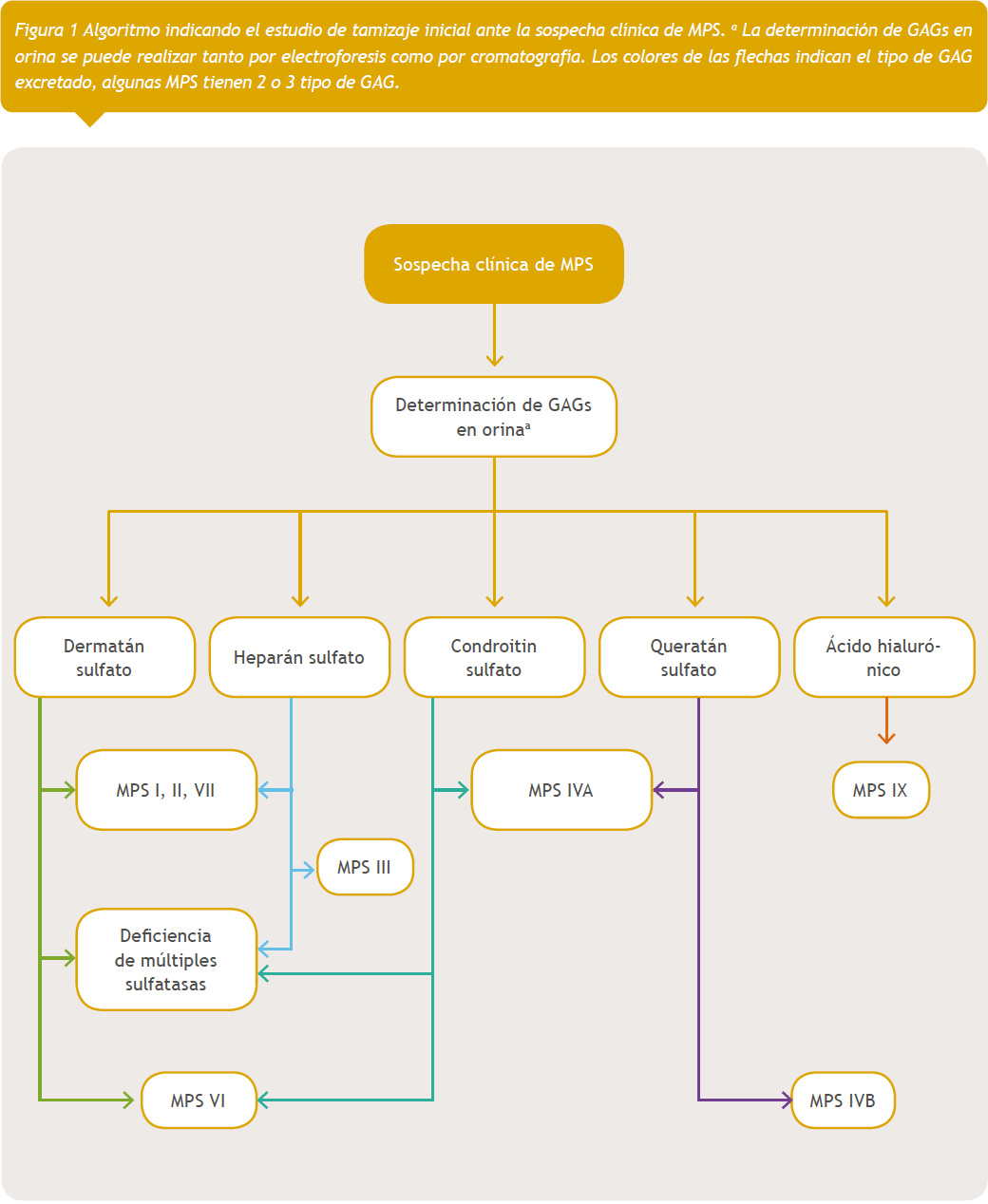
Teniendo en cuenta una sospecha inicial se debe continuar con estudios de tamizaje y posteriormente confirmatorios. Los estudios de tamizaje son iguales para todos los subtipos de MPS, ya que lo que se quiere determinar es si existe excreción de GAG y cuál se excreta en la orina, teniendo la precaución de que a mayor edad disminuye la excreción, y esto no significa que el paciente no tenga la enfermedad. Una vez determinado el tipo GAG en orina se orienta al tipo de ensayo enzimático que se realizará36. La determinación enzimática puede ser realizada en plasma, fibroblastos y leucocitos; se debe tener la precaución de que si se realiza la determinación en plasma siempre debe ser confirmada con determinación en leucocitos o fibroblastos para poder establecer el diagnóstico definitivo. Con este dato confirmatorio se debe iniciar terapia específica para el tipo de MPS demostrado9,19,20,50,51.
Por último, se debe realizar estudio molecular, siempre y cuando esté disponible, para poder establecer el tipo de mutación que presenta, ya que existe correlación genotipo fenotipo en varios casos, y para poder realizar una asesoría genética adecuada buscando portadores en la familia, de tal manera que se pueda prevenir nuevos casos20,50.
En las figuras 1 y 2 se detalla un algoritmo en el diagnóstico de estas entidades desde la sospecha inicial hasta las pruebas confirmatorias por tipo de MPS.
Tratamiento y manejo de las mucopolisacaridosis
El tratamiento de las MPS está dirigido a disminuir la progresión de la enfermedad y mejorar la calidad de vida.
Hasta hace algunos años el tratamiento era paliativo para todos los subtipos, hasta que se iniciaron 2 tipos de tratamientos: el primero de ellos es el trasplante de células madre hematopoyéticas (HSCT) y el segundo la terapia de reemplazo enzimático (ERT).
Para MPS I ha dado buen resultado HSCT de forma temprana; sin embargo, no todos los casos se logran captar tempranamente y deben tratarse con ERT administrada por vía endovenosa, que emplea laronidasa (α–L-iduronidase recombinante humana)5,12,14.
En el caso de MPS II actualmente se emplea para tratamiento ERT con idurosulfasa (I2S) y el HSCT; sin embargo, este último, al contrario de lo que ocurre en MPS I, no ha sido efectivo14,19.
Para MPS III no se ha desarrollado ERT básicamente porque el principal compromiso es del sistema nervioso central, y las enzimas fabricadas hasta la fecha no atraviesan la barrera hematoencefálica. La terapia génica está siendo investigada en modelos animales, al igual que tratamientos con moléculas inhibidoras de síntesis de GAG y estudios en ratones con extractos de isoflavona de soja, los cuales han demostrado efectos gastrointestinales positivos y baja frecuencia de infecciones22.
La enfermedad de Morquio, o MPS IV, tiene tratamiento, que fue aprobado por la FDA, la terapia de reemplazo enzimático es solo para la tipo IV A, la enzima utilizada es la elosulfasa alfa, que corresponde a la enzima recombinante humana N acetilagalactosamina-6-sulfatasa52.
Para MPS VI la ERT se hace con galsulfasa, una forma recombinante de la enzima N-acetilgalactosamina 4-sulfatasa14. Otra de las opciones terapéuticas es HSCT14,38, e incluso se ha propuesto la combinación de la ERT y HSCT; sin embargo, se considera la ERT como tratamiento principal14.
Para las otras formas de MPS no existe hasta la fecha ERT, pero se debe precisar que el manejo de cada una de estas no solo es la ERT, sino que consiste en un manejo transdisciplinario, con la participación de cada uno de los especialistas de acuerdo a las áreas de compromiso. Para cada subtipo de MPS existen guías de manejo preventivo y de las complicaciones que se puedan presentar.
Conclusiones
Las MPS son un amplio grupo de patologías infrecuentes, pero con impacto para el paciente, la familia y la sociedad muy alto. Por esta razón es importante reconocer sus características para poder establecer un diagnóstico oportuno y ofrecer un tratamiento adecuado, teniendo en cuenta adicionalmente que existen para cuatro de estas enfermedades ERT y su inicio temprano sumado al manejo transdisciplinario mejora la calidad de vida y el pronóstico.
Referencias Bibliográficas
1. Bouzidi H., Khedhiri S., Laradi S., Ferchichi S., Daudon M., Miled A. La mucopolysaccharidosis IVA (syndrome de Morquio A): Aspects clinique, biologique et thérapeutique. Ann Biol Clin (Paris). 2007;65:5-11.
2. González-Meneses A., Barcia A., Di¿az J.L. Protocolo de actuación en las mucopolisacaridosis. Protoc Diagn Ter Pediatr. 2010;1:24-36.
3. Henderson J.L. Gargoylism: A review of the principal features with a report of five cases. Arch Dis Child. 1940;15:201-14.
4. Hunter C. A rare disease in two brothers. Proc R Soc Med. 1917;10:104-16.
5. Bay L., Amartino H., Barreiro C., et al. Consenso de diagnóstico y tratamiento de la mucopolisacaridosis de tipo I. Arch Argent Pediatr. 2008; 106:361-8.
6. Chudley A.E., Chakravorty C. Genetic landmarks through philately: Luis Morquio 1867-1935. Clin Genet. 2002;62:438-9.
7. Cunningham R. A contribution to the genetics of gargoylism. J Neurol Neurosurg Psychiatry. 1954;17:191-5.
8. Al-Maawali A., Surendra J., Roshan K. Spectrum of paediatric lysosomal storage disorders in Oman. SQU Med J. 2012;12:295-9.
9. Uribe A., Giugliani R. Selective screening for lysosomal storage diseases with dried blood spots collected on filter paper in 4,700 high-risk Colombian subjects. JIMD. 2013;11:109-16.
10. Natowicz M., Short M., Wang Y., et al. Clinical and biochemical manifestations of hyaluronidase deficiency. N Engl J Med. 1996;335:1029-33.
11. McKusick V.A. The nosology of the mucopolysaccharidoses. Am J Med. 1969;47:730-47.
12. D’Aco K., Underhill L., Rangachari L., et al. Diagnosis and treatment trends in mucopolysaccharidosis I: Findings from the MPS I Registry. Eur J Pediatr. 2012;171:911-9.
13. De M., Teunissen Q., van J., et al. Capturing phenotypic heterogeneity in MPS I: Results of an international consensus procedure. Orphanet J Rare Dis. 2012;7:22.
14. Giugliani R., Federhen A., Rojas M., et al. Mucopolysaccharidosis I, II, and VI: Brief review and guidelines for treatment. Genet Mol Biol. 2010;33:589-604.
15. Munoz-Rojas M., Bay L., Sanchez L., et al. Clinical manifestations and treatment of mucopolysaccharidosis type I patients in Latin America as compared with the rest of the world. J Inherit Metab Dis. 2011;34:1029-37.
16. De M.H., Boelens J., Das A., et al. Enzyme replacement therapy and/or hematopoietic stem cell transplantation at diagnosis in patients with mucopolysaccharidosis type I: Results of a European consensus procedure. Orphanet J Rare Dis. 2011;6:55.
17. Menéndez-Sainz C., González-García S., Zaldívar-Muñoz C., González-Quevedo A. Mucopolisacaridosis con afectaciones del sistema nervioso. Rev Mex Neuroci. 2006;7:150-5.
18. Manger B., Mengel E., Schaefer R. Rheumatologic aspects of lysosomal storage diseases. Clin Rheumatol. 2007;26:335-41.
19. Gullebart N., Amartino H., Arberas C. Guía para el diagnóstico, seguimiento y tratamiento de la mucopolisacaridosis de tipo II (MPS-II) o Enfermedad de Hunter. Argent Pediatr. 2011;2:175-81.
20. Wraith J., Scarpa M., Beck M., Bodamer O., et al. Mucopolysaccharidosis type II (Hunter syndrome): A clinical review and recommendations for treatment in the era of enzyme replacement therapy. Eur J Pediatr. 2008;167:267-77.
21. Martin R., Beck M., Eng C., et al. Recognition and diagnosis of mucopolysaccharidosis II (Hunter syndrome). Pediatrics. 2008;121:e377-86.
22. Wijburg F., Wegrzyn G., Burton B., Tylki-Szymanska A. Mucopolysaccharidosis type III (Sanfilippo syndrome) and misdiagnosis of idiopathic developmental delay, attention deficit/hyperactivity disorder or autism spectrum disorder. Acta Paediatr. 2013;102:462-70.
23. Cleary M., Wraith J. Management of mucopolysaccharidosis type III. Arch Dis Child. 1993;69:403-5.
24. Esposito S., Balzano N., Daniele A., et al. Heparan N-sulfatase gene: Two novel mutations and transient expression of 15 defects. Biochim Biophys Acta. 2000;1501:1-11[Links]
25. Wolfea B., Ghomashchi F., Kima T., et al. New substrates and enzyme assays for the detection of mucopolysaccharidosis III (Sanfilippo syndrome) Types A, B, C and D by tandem mass spectrometry. Bioconjug Chem. 2012;23:557-64. [Links]
26. Burrows R., Muzzo S. Síndrome de San Filippo: tipificación de mucopolisacaridosis en orina y determinación enzimática en plasma. Rev Chil Pediatr. 1980;50:129-33. [Links]
27. Suárez-Obando F., Zarante-Montoya I. Aspectos clínicos y manejo integral del síndrome de Morquio. Univ Méd. 2007;48:166-74. [Links]
28. Baker E., Guo X.H., Orsborn A.M., et al. The Morquio A syndrome (mucopolysaccharidosis IVA) gene maps to 16q24.3. Am J Hum Genet. 1993;52:96-8. [Links]
29. Maceira-Rozas M.C., Atienza-Merino G. Deteccio¿n precoz de mucopolisacaridosis y oligosacaridosis en el periodo neonatal mediante cribado poblacional. Revisión sistemática. Madrid: Ministerio de Sanidad y Consumo, (2006) pp. 21-124. [Links]
30. The Gene Ontology. N-acetylgalactosamine-6-sulfatase activity; 2013 [consultado 14 Jun 2014]. Disponible en:http://amigo.geneontology.org/cgi-bin/amigo/term_details?term=GO:0043890#top. [Links]
31. Caciotti A., Garman S.C., Rivera-Colon Y., et al. GM1 gangliosidosis and Morquio B disease: An update on genetic alterations and clinical findings. Biochim Biophys Acta. 2011;1812:782-90. [Links]
32. Northover H., Cowie R.A., Wraith J.E. Mucopolysaccharidosis type IVA (Morquio syndrome): A clinical review. J Inherit Metab Dis. 1996;19:357-65. [Links]
33. Mena M., Obando R. Síndrome de Morquio. Rev Chil Pediatr. 1976;47:247-54. [Links]
34. Suarez-Guerrero J.L., Bello-Suárez A.K., Vargas-Santos M.C., Contreras-Garcia G.A. Caracterización clínica, estudios genéticos y manejo de la mucopolisacaridosis tipo IVA. MÉD UIS. 2013;26:43-50. [Links]
35. Amigo O., Burrows R., Muzzo S. Mucopolisacaridos tipo iv. Síndrome de Morquio. Rev Chil Pediatr. 1979;50:61-4. [Links]
36. Chamoles N.A., Blanco M.B., Gaggioli D., Casentini C. Hurler-like phenotype: Enzymatic diagnosis in dried blood spots on filter paper. Clin Chem. 2001;47:2098-102. [Links]
37. Tomatsu S., Montano A.M., Oikawa H., et al. Mucopolysaccharidosis type IVA (Morquio A disease): Clinical review and current treatment: A special review. Curr Pharm Biotechnol. 2011;12:931-45. [Links]
38. Valayannopoulos V., Nicely H., Harmatz P., Turbeville S. Mucopolysaccharidosis VI. Orphanet J Rare Dis. 2010;5:5. [Links]
39. Harmatz P. Enzyme replacement therapy with galsulfase for mucopolysaccharidosis VI: Clinical facts and figures. Turk J Pediatr. 2010;52:443-9. [Links]
40. Litjens T., Hopwood J.J. Mucopolysaccharidosis type VI: Structural and clinical implications of mutations in N-acetylgalactosamine-4-sulfatase. Hum Mutat. 2001;18:282-95. [Links]
41. Braunlin E., Rosenfeld H., Kampmann C., et al. Enzyme replacement therapy for mucopolysaccharidosis VI: Long-term cardiac effects of galsulfase (Naglazyme®) therapy. J Inherit Metab Dis. 2013;36:385-94. [Links]
42. Sly W.S., Quinton B.A., McAlister W.H., Rimoin D.L. Beta glucuronidase deficiency: Report of clinical, radiologic, and biochemical features of a new mucopolysaccharidosis. J Pediatr. 1973;82:249-57. [Links]
43. Cheng Y., Verp M.S., Knutel T., Hibbard J.U. Mucopolysaccharidosis type VII as a cause of recurrent non-immune hydrops fetalis. J Perinat Med. 2003;31:535-7. [Links]
44. Tomatsu S., Montaño A.M., Dung V.C., Grubb J.H., Sly W.S. Mutations and polymorphisms in GUSB gene in mucopolysaccharidosis VII (Sly syndrome). Hum Mutat. 2009;30:511-9. [Links]
45. Yamada Y., Kato K., Sukegawa K., et al. Treatment of MPS VII (Sly disease) by allogeneic BMT in a female with homozygous A619 V mutation. Bone Marrow Transplant. 1998;21:629-34. [Links]
46. Artigalás O.A., da L.R., Burin M., et al. Multiple sulfatase deficiency: Clinical report and description of two novel mutations in a Brazilian patient. Metab Brain Dis. 2009;24:493-500. [Links]
47. Basner R., von K., Glössl J., Klein U., Kresse H., Mlekusch W. Multiple deficiency of mucopolysaccharide sulfatases in mucosulfatidosis. Pediatr Res. 1979;13:1316-8. [Links]
48. Blanco-Aguirre M.E., Kofman-Alfaro S.H., Rivera-Vega M.R., et al. Unusual clinical presentation in two cases of multiple sulfatase deficiency. Pediatr Dermatol. 2001;18:388-92. [Links]
49. Burk R.D., Valle D., Thomas G.H., et al. Early manifestations of multiple sulfatase deficiency. J Pediatr. 1984;104:574-8. [Links]
50. Wood T.C., Harvey K., Beck M. Diagnosing mucopolysaccharidosis IVA. J Inherit Metab Dis. 2013;36:293-307. [Links]
51. Scarpa M., Almássy Z., Beck M. Mucopolysaccharidosis type II: European recommendations for the diagnosis and multidisciplinary management of a rare disease. Orphanet J Rare Dis. 2011;7:72. [Links]
52. Tomatsu S., Sawamoto K., Alméciga-Díaz C.J., et al. Impact of enzyme replacement therapy and hematopoietic stem cell transplantation in patients with Morquio A syndrome. Drug Des Devel Ther. 2015;9:1937-53. [Links]
Conflicto de intereses
Este trabajo cumple con los requisitos sobre consentimiento/asentimiento informado, comité de ética, financiación, estudios animales y sobre la ausencia de conflicto de intereses según corresponda.
Autor para correspondencia.


Más notas de la edición 91
Lee desde Issuu nuestra última edición publicada en Octubre 2025, Edición número 170
Notas relacionadas a Mucopolisacaridosis: características clínicas,...


