
Hipotiroidismo congénito: un diagnóstico que no debemos olvidar
Francisca Grob L.1, Alejandro Martínez-Aguayo1
1.División de Pediatría, Unidad de Endocrinología Pediátrica, Pontificia Universidad Católica de Chile.
Rev Chil Pediatr 2012; 83 (5): 482-491
Artículo de Revisión
Correspondencia: Alejandro Martínez- Aguayo E-mail: alemarti@med.puc.cl
Alcalde Eduardo Castillo Velasco 1838, Ñuñoa, Santiago. Casilla 593-11.
Tel.: (56-2) 2237 1598 -2237 9757
Fax: (56-2) 2238 0046
revista@sochipe.cl – Rev Chil Pediatr 2012; 83 (5): 482-491
Abstract
Summary: Congenital hypothyroidism (CH) is the most common cause of preventable mental retardation. Since 1994, Chile has a national plan for mass screening all newborns to diagnose the disease. Currently, the CH incidence is approximately 1:3163 newborn (NB). Approximately, 10% of these cannot be identified by screening programs, so the clinical suspicion is fundamental in the diagnosis. The most frequently clinical features observed in neonates or young infants are the presence of a posterior fontanelle greater than 5 mm, umbilical hernia and dry skin. It is important to determine the etiology of CH, but the etiological study should not delay the start of treatment. Early treatment determines a better prognosis of neurological development. A review of the CH screening program, pathophysiology, clinical presentation, and aspects of the study and treatment are presented in this study.
Key words: Congenital hypothyroidism, thyroid dysgenesis, neonatal screening, false negative, neurological development.
Resumen
El hipotiroidismo congénito (HTC) es la causa más frecuente de discapacidad intelectual prevenible. Desde el año 1994 existe en Chile un plan nacional de tamizaje masivo a todos los recién nacidos para el diagnóstico de la enfermedad. Actualmente, la incidencia de HTC es de aproximadamente 1:3 163 recién nacidos (RN). Hasta un 10% de éstos puede no ser identificado por los programas de tamizaje, por lo que es importante la sospecha clínica del diagnóstico. Las características clínicas más frecuentemente observadas en RN o lactantes pequeños son la presencia de una fontanela posterior mayor de 5 mm, hernia umbilical y piel seca. Es importante determinar la etiología del HTC, pero el estudio etiológico no debe retrasar el inicio del tratamiento. El inicio precoz de éste determina un mejor pronóstico de desarrollo neurológico. Se presenta una revisión del programa de tamizaje de HTC, su fisiopatología, presentación clínica, y aspectos del estudio y tratamiento.
Palabras clave: Hipotiroidismo congénito, Disgenesia Tiroidea, Tamizaje neonatal, Falso negativo, desarrollo neurológico.
Introducción
El hipotiroidismo congénito (HTC) es la endocrinopatía y causa de discapacidad cognitiva prevenible más frecuente en el recién nacido. El pronóstico del desarrollo neurológico se relaciona en forma inversa a la edad de diagnóstico e inicio de tratamiento de la enfermedad. Por esta razón, se han desarrollado programas de tamizaje neonatal en todo el mundo para la detección oportuna de esta patología.
El primer programa de tamizaje para la detección de HTC se desarrolló en Canadá el año 1973, empleando la estrategia de medición de T4 en los recién nacidos1. En Latinoamérica, Cuba fue pionero en iniciar un programa nacional de tamizaje masivo, seguidos por Costa Rica, Chile y Uruguay. El programa en estos países se caracteriza por su manejo a nivel nacional y cobertura cercana al 100% de la población. Actualmente, aun existen países de nuestra región en que la cobertura es inferior al 1%, como Guatemala, República Dominicana, Bolivia, Panamá y Ecuador. En Chile, el Ministerio de Salud desarrolló el programa de tamizaje neonatal para HTC a partir del año 1992 para evaluar a todos recién nacidos, alcanzando un 100% de cobertura el año 1982. Este programa consiste en determinar niveles de TSH en una muestra de sangre total obtenida en una tarjeta de papel filtro, a partir de las 40 h de vida en los recién nacidos de término sobre 37 semanas de edad gestacional (EG), en los recién nacidos de pretérmino (RNPT) entre 35 a 36 semanas de EG a los 7 días de vida, y en los RNPT menores de 35 semanas de EG a los 7 días de vida, y repetir una muestra a los 15 días de vida. Los resultados se envían a los laboratorios de referencia (Hospital San Juan de Dios; Instituto Nacional de Tecnología en Alimentos INTA; Hospital Guillermo Grant Benavente, Concepción) donde se procesa la muestra. Algunas clínicas privadas procesan las muestras de sus recién nacidos en sus propios laboratorios.
Antes de la introducción del tamizaje neonatal, se estima que la incidencia de HTC era entre 1:7 000 a 1:10 000 en el mundo. Esta difiere en los diferentes países de acuerdo al método y el punto de corte de tamizaje de TSH utilizado para la detección de la patología, además de otros factores. Es así como en Latinoamérica la mayor incidencia de HTC se presenta en Paraguay (1:1 667) y la menor en Brasil ( 1:3 670)3. En Chile, la incidencia de HTC se estima en 1:3 163. Entre marzo de 1992 y diciembre de 2008, el diagnóstico de HTC fue confirmado a una edad promedio de 12,5 ± 6,9 días, con un valor promedio de TSH de tamizaje de 218,7 ± 31,8 mU/mL. Los falsos positivos alcanzaron el 1%, dado principalmente por toma precoz de la muestra2.
La estrategia de tamizaje neonatal utilizada en Chile es la determinación de TSH. Se utiliza un punto de corte en sangre en papel filtro de TSH 15 uIU/mL con el método de inmunofluorescencia a tiempo retardado (DEL-FIA). Esto quiere decir que valores de TSH iguales o mayores a 15 uIU/mL obligan a confirmar el diagnóstico de HC en una muestra de sangre venosa. Antiguamente, se determinaba la TSH a través del método radioinmunométrico (IRMA), que por el hecho de ser menos sensible, utiliza un punto de corte de 20 mUI/ mL. La estrategia de tamizaje con medición de TSH no detecta variantes de hipotiroidismo congénito caracterizados por presentarse con concentraciones bajas de TSH, como el hipotiroidismo central, el déficit de la proteína de unión de tiroxina (TBG), la hipotiroxinemia y el hipotiroidismo del RNPT. En los RNPT, la presencia de un eje hipotálamo hipófisis tiroideo inmaduro determina elevaciones más tardías de la TSH, además, el uso habitual de dopamina por patologías concomitantes puede atenuar aún más su elevación4. Los niños con hipotiroidismo congénito atípico, caracterizados por una elevación tardía de TSH, tampoco serán detectados inicialmente. Por lo tanto, debe sospecharse hipotiroidismo en todo niño con síntomas y/o signos característicos de la enfermedad, aún así haya presentado un tamizaje neonatal negativo.
El tamizaje neonatal de HTC presenta falsos negativos y falsos positivos. se ha estimado que entre un 5 a 10% de los recién nacidos pueden no ser detectados por los programas de tamizaje (denominados falsos negativos)5. Las causas pueden estar relacionadas con factores biológicos (hipotiroidismo central, uso de productos yodados en la madre y/o hijo) o errores en el tamizaje (falla en la recolección de la muestra, muestras insatisfactorias, error en la interpretación del resultado y/o falla en el rellamado), por lo que la recomendación es que el tamizaje se lleve a cabo en laboratorios que participen en programas internacionales de control. Los falsos positivos pueden presentarse cuando el tamizaje se realiza antes de las 48 h de vida (por la elevación fisiológica de la TSH durante las primeras horas de vida), y cuando el punto de corte de TSH de tamizaje para el diagnóstico es menor. Esta estrategia mejora la sensibilidad del método, pero aumenta la tasa de rellamado, aumentando los costos emocionales y económicos para la familia y el programa6.
Al analizar la relación costo efectividad de realizar tamizaje de HTC, se ha determinado que los programas representan un ahorro económico neto para la sociedad. Para calcular, se han considerado factores tales como los costos asociados a la discapacidad intelectual, la expectativa de vida de la población, los costos del programa y del tratamiento a largo plazo7,8. El programa previene en forma sustancial la discapacidad intelectual secundaria a HTC, por lo que el programa justifica su búsqueda masiva9.
Embriología de la glándula tiroides
El desarrollo de la glándula tiroides se inicia a partir de la cuarta semana de desarrollo embrionario y se encuentra regulado por una serie de genes. El tiroides se origina como engrosamiento del endodermo en la pared ventral de la faringe primitiva, caudal a la región del primer arco branquial, proceso denominado especificación”. Luego, el primordio tiroideo comienza a invadir el mesénquima circundante, formando un brote que prolifera y migra desde el piso faríngeo hacia su ubicación definitiva, la porción media anterior del cuello en la séptima semana del desarrollo. En la octava semana, se identifica un pequeño istmo, y dos lóbulos laterales. Un error en este período produce alteraciones anatómicas del tiroides, es decir, disembriogénesis o disgenesia tiroidea (aplasia, hipoplasia o ectopia tiroidea). Hasta en un 2% de estos pacientes se han identificado mutaciones en los factores de transcripción PAX8, NKX2-1, FOXE1, NKX2-510; se deduce que la mayoría de los genes implicados en el desarrollo tiroideo aun no han sido identificados. Mutaciones en éstos factores se han asociado con otras manifestaciones clínicas (tabla 1).
Tabla I. Mutaciones de factores de transcripción y genes que producen disembriogenesis y dishormonogénesis y sus características clínicas asociadas
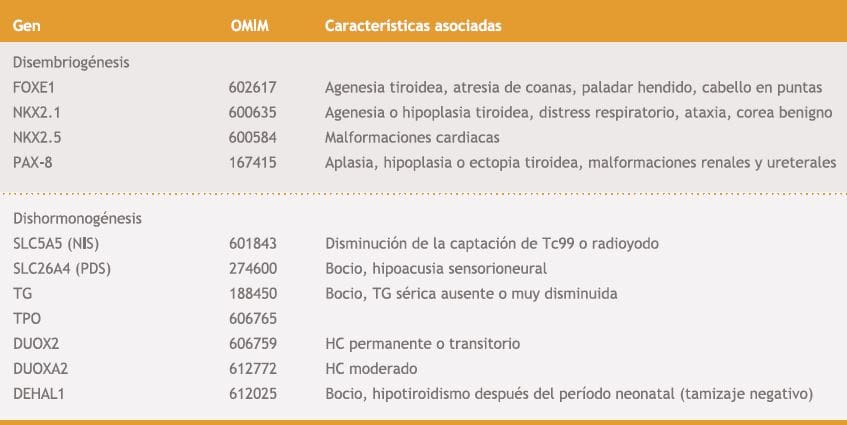
NIS: Sodium iodide symporter, PDS: Pendred syndrome, TG: Tiroglobulina, TPO: thyroid peroxidase, DUOX2: Dual oxidase 2, DUOXA2: DUOX maturation factor 2, DEHAL 1: Deyodinasa.
Una vez ubicada en la porción anterior del cuello (10 a 12 semanas del desarrollo) se inicia el período de diferenciación funcional de las células foliculares y comienza la expresión de proteínas necesarias para la síntesis de hormonas tiroideas (Transportador sodio- yodo, Pendrina, Tiroglobulina, Peroxidasa tiroídea, Receptor de TSH, Oxidasas tiroideas) (figura 1). Una alteración de éstas resulta en dishor-monogénesis (OMIM # 274400), es decir, alteraciones en la síntesis de hormonas tiroideas, con un tiroides de ubicación normal.
Figura I. Síntesis de hormonas tiroideas en el tirocito.
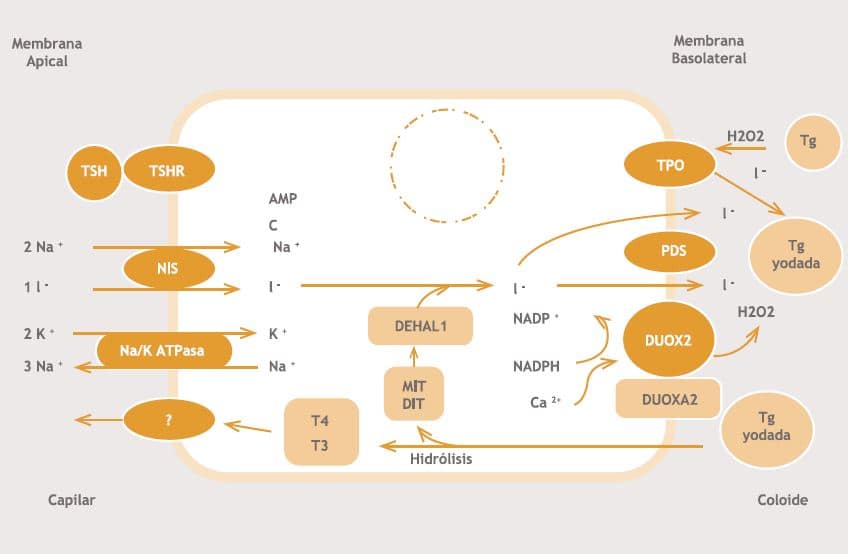
1) El yodo inorgánico es transportado activamente a través de la membrana basolateral de los tirocitos, a través del cotransportador de sodio-yodo (NIS); 2) Luego, desde de la membrana apical hacia el lumen folicular a través de Pendrina (PDS); 3) En la superficie apical de los tirocitos, el yodo es oxidado por la pe-roxidasa tiroidea (TPO) y 4) Se incorpora a los residuos de tirosina de la tiroglobulina (Tg) para formar monoyodotirosinas y diyodotirosinas. TPO también cataliza la unión de las yodotirosinas en yodotironinas. La yodación de la Tg requiere peróxido de hidrógeno (H2O2), el que es producido por dos oxidasas (DUOXs) de manera dependiente de NADPH. Factores DUOX de maduración (DUOXAs) están involucrados en el tráfico de membrana de DUOXs. La Tg yodada es internalizada al tirocito por pinocitosis, y se somete a la degradación proteolítica en el lisosoma para liberar T3 y T4.

Fisiología
La producción y secreción de hormonas tiroideas está regulada por el eje hipotálamo-hipófisis-tiroides (HHT) (figura 2). El eje fetal es funcional al final de primer trimestre del embarazo. Durante el primer trimestre, el desarrollo neurológico del feto dependerá de adecuadas concentraciones de T3 y T4 maternas, las que traspasan la placenta y protegen al feto de hipotiroidismo. Niños con ausencia de tiroides, presentan niveles de T4 al momento de nacer entre 20 a 50% de la concentración de un recién nacido normal, esto se explica por el paso transplacentario de hormonas tiroideas maternas. El hipotálamo secreta la hormona liberadora de tirotropina (TRH) que estimula a los tirotropos hipofisiarios para que secreten TSH. Ésta estimula el crecimiento tiroideo, la síntesis y secreción fundamentalmente de tiroxina (T4), la que se convierte en su forma fisiológicamente más activa, la T3, gracias a las deyodinasas ubicadas en los tejidos periféricos. Las hormonas tiroideas circulan en la periferia unidas a proteínas, predominantemente la thyroid binding globuline (TBG), también a transtiretina y albúmina. En los tejidos periféricos, las hormonas tiroideas actúan uniéndose a receptores específicos en el DNA (TRa1, TRp1, TRp2, TRp3). El hipotiroidismo puede resultar de una alteración en cualquier de estos niveles.
Figura 2. Eje Hipotálamo-Hipófisis-Tiroides (HHT) y acción periférica de las hormonas tiroideas (HT).
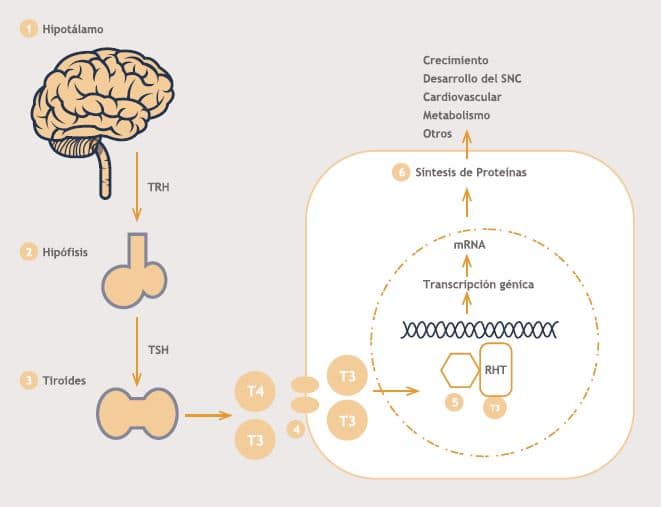
TRH: Hormona liberadora de tirotropina, TSH: Hormona estimulante tiroidea, RHT: Receptor de hormonas tiroideas. (1) El hipotálamo a través de la secreción de TRH estimula la síntesis de TSH hipo-fisiaria (2), la que a su vez estimula el tiroides para la producción de T3 y T4 (3). Éstas ingresan a la célula a través de un transportador de membrana (4), y luego al núcleo, donde interactúan con el RHT para regular la transcripción de genes target (5). Estos genes son responsables de la síntesis de proteínas (6) necesarias para una serie de funciones.
Etiología
Las etiologías incluyen el HTC primario (disembriogénesis y dishormonogénesis), hipotiroidismo central e hipotiroidismo transitorio (inducido por yodo, drogas o anticuerpos maternos) (tabla 2). La causa más frecuente de HC es la disembriogénesis tiroidea, que corresponde al 85% del total de casos. La ectopia es la causa más frecuente de disembriogénesis (2/3 de los casos) seguidos de aplasia e hipoplasia tiroidea11. Las dishormonogénesis representan la segunda causa de HC (15% de los casos), se heredan en forma autosómica recesiva, y se producen por mutaciones en las proteínas responsables del transporte, oxidación y organificación del yodo (tabla 2). Dentro de las causas de hipotiroidismo transitorio, se debe descartar la presencia de anticuerpos maternos (que bloquean el receptor de TSH neonatal), el déficit o exceso de yodo y el uso de drogas anti tiroídeas maternas.
Manifestaciones clínicas
Las características clásicas de HC casi no se observan hoy en día en los países que cuentan con programas de tamizaje neonatal, es por esto que la sospecha clínica debe ser aún mayor. Se estima que sólo un 25% de los niños nacen en países que cuentan con programas de tamizaje neonatal12. En el resto, el diagnóstico debe ser sospechado por la clínica (tabla 3), sin embargo, ésta es poco sensible para el diagnóstico precoz de la patología. Un estudio en Dinamarca demostró que sólo 10% de los afectados fueron diagnosticados durante el primer mes de vida, 35% los primeros 3 meses y 70% durante el primer año de vida. En el resto, el diagnóstico se retrasó al tercer y cuarto año de vida13. Un análisis retrospectivo de 1000 casos de HTC de Turquía determinó que la edad media al diagnóstico fue de 49 meses y sólo el 3,1% de los casos fueron diagnosticados en el primer mes, mientras que el 55,4% fueron diagnosticados después de los 2 años14.

Tabla III. Características clínicas de hipotiroidismo congénito
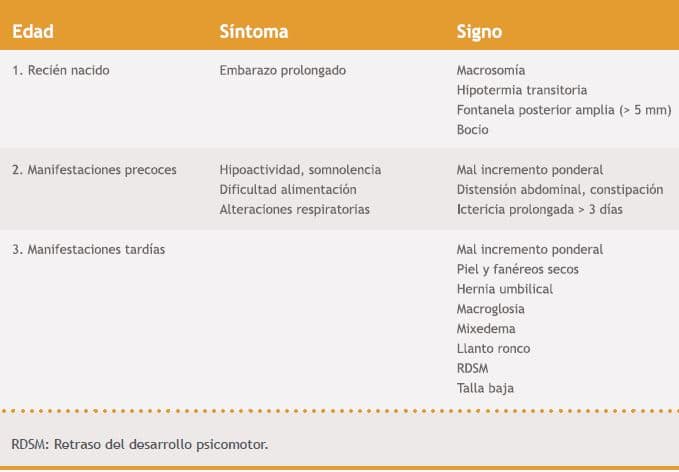
La razón por la que los síntomas de HTC pueden ser sutiles al momento de nacer, incluso en pacientes con hipotiroidismo bioquímico severo, radica en el paso de hormonas tiroideas maternas a través de la placenta durante el desarrollo neurológico fetal lo protege de manifestaciones iniciales importantes. Dentro de los antecedentes, hasta en el 20% puede existir la historia de un embarazo prolongado15. Al nacer, el peso y longitud de nacimiento habitualmente son normales, pueden presentar una circunferencia craneana mayor. Si el HTC no es pesquisado por el tamizaje, durante los 3 primeros meses de vida aparecerá letargia, hipotonía, aumento del tamaño de la lengua, llanto ronco, hernia umbilical, piel seca y moteada, constipación e ictericia prolongada. La presencia de bocio puede manifestarse al momento del nacimiento o más tarde en la infancia si la causa es la dishormonogénesis16.
En Hungría, evaluaron síntomas y signos de hipotiroidismo al momento del rellamado en recién nacidos con tamizaje positivo. Compararon los s síntomas y signos de los niños que presentaron una confirmación positiva con aquellos que presentaron una confirmación negativa al momento de la confirmación17, y se observó que en este momento aquellos con confirmación positiva ya presentaban una fontanela posterior mayor de 5 mm (78% vs 20%), hipoactividad (67% vs 40%), piel seca (72% vs 35%) e ictericia (55% vs 20%). Los síntomas y signos se presentan con mayor frecuencia mientras más profundo sea el HTC. Al comparar la clínica al momento del diagnóstico utilizando un punto de corte de T4 total de 30 nmol/L, aquellos con T4 total menor o igual a 30 nmol/L, presentaban con mayor frecuencia ictericia prolongada (59% vs 33%), dificultad para alimentarse (35% vs 16%), letargia (34% vs 14%), hernia umbilical (32% vs 18%) y macroglosia (25% vs 12%)18. Además, presentan con mayor frecuencia comorbilidades perinatales (16% vs 9%). La prevalencia de malformaciones congénitas asociadas, independiente de la causa, es mayor que la de la población general. Ésta varía entre un 7 a 23%, dependiendo de la población estudiada siendo las más frecuentes las cardiopatías congénitas19
Evaluación diagnóstica
Existen 3 estrategias de tamizaje20: 1) T4 inicial, si el resultado se encuentra bajo el punto de corte, se realiza TSH; 2) TSH inicial y 3) TSH y T4 inicial combinados. En Chile se utiliza la estrategia de TSH inicial. Cada programa de tamizaje establece sus propios puntos de corte, pero habitualmente si la T4 de tamizaje se encuentra bajo el percentil 10 y/o la TSH es mayor a 15 mU/L en sangre total, se solicita la confirmación (figura 3). Frente a una prueba de tamizaje alterada, los recién nacidos deben ser contactados en forma inmediata para ser evaluados y confirmar el resultado midiendo TSH y T4 total en una muestra de sangre venosa. Idealmente se debe preferir medir niveles de T4 libre antes que T4 total: la determinación de T4 total obliga a determinar además TBG para descartar un déficit de esta proteína cuando la TSH es normal con T4 total baja. Si la TSH es superior a 10 uIU/mL y la T4 es inferior a 10 ug/dl se confirma el diagnóstico de HC.
Figura III. Tamizaje de Hipotiroidismo congénito en recién nacidos. Adaptado con permiso de Macmillan Publishers Ltd: Nature reviews in endocrinology. Grüters A, Krude H. Detection and treatment of congenital hypothyroi-dism. Nat Rev Endocrinol. 2011 Oct 18;8(2):104-13. Copyright 2011.


En relación al estudio de imágenes, una vez confirmado el diagnóstico, se pueden solicitar estudios adicionales para determinar la etiología del HC, siempre y cuando no se retrase el inicio precoz del tratamiento. La ecografía tiroídea permitirá definir el subgrupo de HC (disembriogénesis o dishormonogénesis) frente a la presencia o ausencia de glándula tiroides. Ésta debe ser realizada por un radiólogo pediatra experto, con un equipo de alta resolución. De estos factores dependerá la sensibilidad del método, especialmente para la detección de tejido tiroideo ectópico21
El cintigrama tiroídeo con tecnecio 99 permitirá definir la localización exacta de una glándula ectópica. Este puede realizarse al momento del diagnóstico, incluso en etapas iniciales del tratamiento puesto que éste no normaliza en forma inmediata la TSH. Si la TSH ya ha descendido a valores inferiores de 30 mU/L, se puede realizar una ecografía tiroidea20. Si no existe disponibilidad de cintigrama, éste puede diferirse hasta la edad de 3 años en que se suspende el tratamiento con levotiroxina y se reevalua. La ausencia de captación por el cintigrama con una glándula pequeña de localización normal, puede ser explicada por mutaciones del receptor de TSH, defectos del atrapamiento del yodo (NIS) y presencia de anticuerpos estimuladores del receptor de TSH (TRAB) maternos.
La tiroglobulina (TG) es un marcador específico de tejido tiroideo y permite diferenciar los casos con cintigrama no captante. Frente a TG ausente, se confirma la aplasia tiroidea. Si ésta se encuentra elevada, plantearemos la presencia de tejido tiroideo, pero que por algún defecto en los receptores de membrana tiroidea (mutación de receptor de NIS o de TSH u ocupación de éste por TRAB maternos inactivantes), ésta no es detectada ya que el radiofármaco no ingresa al tiroides. En el caso de las dishormonogénesis (HC con tiroides normotópico), con concentraciones bajas de TG orienta a mutaciones de ésta proteína. El test de descarga con perclorato se utiliza para diagnosticar defectos de organificación (mutaciones en los genes TPO, DUOX2 y DUOXA2). Se realiza un cintigrama con yodo 123, luego se administra perclorato de sodio y se determina la captación tiroidea. Una descarga es superior al 50% indica un defecto de organificación virtualmente total.
Para descartar déficit o exceso de yodo, se puede determinar yodo en orina (en neonatos, entre 50 a 100 ug/24 h). La yoduria estará disminuída en áreas endémicas de déficit de yodo, y elevada en casos de ingestión excesiva de la madre o uso de antisépticos yodados en la madre o recién nacido periparto. En niños con hipotiroidismo bioquímico y glándula tiroides normotópica, se debe descartar además paso de anticuerpos maternos (los anticuerpos involucrados pueden ser los anticuerpos antitiroglobulina y antiperoxidasa, o los anticuerpos bloqueadores del receptor de TSH, esta causa representa el 2% del total de hipotiroidismos congénitos y puede producirse sólo si la madre presenta una enfermedad tiroidea autoinmune)22 y sobrecarga o déficit de yodo23.
Tratamiento
El tratamiento con hormonas tiroideas debe iniciarse una vez obtenida la muestra de confirmación, sin esperar su resultado ni el de los exámenes complementarios. Del inicio oportuno del tratamiento, la dosis y la monitorización de la terapia dependerá la evolución del desarrollo neurológico de los niños afectados por HTC.

El objetivo del tratamiento es normalizar los niveles de T4 en 2 semanas y los de TSH en un mes. La administración de levotiroxina es el tratamiento de elección. El comprimido debe ser triturado y disuelto en leche materna, fórmula o agua. Respecto a la dosis a utilizar, las guías chilenas24, y Norteamericanas20 sugieren iniciar el tratamiento con 10 a 15 ug/kg/ día de levotiroxina apenas se haya tomado la muestra confirmatoria. Sólo un estudio randomizado ha determinado que RNT que iniciaron tratamiento con dosis mayores de levotiroxina (50 ug/día) presentaron resultados significativamente mejores en pruebas de inteligencia que aquellos niños que recibieron dosis habituales (10 a 15 ug/kg/ día)25. La evidencia hasta el momento es insuficiente para sugerir que iniciar el tratamiento con dosis altas de levo-tiroxina es más beneficioso que dosis bajas26, pero probablemente lo más adecuado sea iniciar el tratamiento con dosis más elevadas (50 ug/día) para normalizar rápidamente los niveles de T4.
Conclusiones
Presentamos una revisión del tamizaje de hipotiroidismo congénito y el diagnóstico clínico precoz de esta patología. Nos parece importante recalcar que el tamizaje neonatal presenta hasta un 10% de falsos negativos. Actualmente en Chile no está reportada la incidencia de falsos negativos. Es por esta razón que la sospecha clínica de hipotiroidismo congénito debe mantenerse en niños que presenten síntomas y/o signos de la enfermedad.
Referencias Bibliográficas.
1.- Dussault JH, Laberge C. Thyroxine (T4) determination by radioimmunological method in dried blood eluate: new diagnostic method of neonatal hypothyroidism?. Union Med Can 1973; 102 (10): 2062-4.
2.- Cornejo V, Raimann E, Cabello JF. Past, present and future of newborn screening in Chile. J Inherit Metab Dis 2010;00:0-0.
3.- Borrajo GJ. Newborn screening in Latin America at the beginning of the 21st century. J Inherit Metab Dis 2007; 30 (4): 466-81.
4.- Filippi L, Pezzati M, Cecchi A, Poggi C. Dopamine infusion: a possible cause of undiagnosed congenital hypothyroidism in preterm infants. Pediatr
Crit Care Med 2006; 7 (3): 249-51.
5.- Yunis KA, Nasr MR, Lepejian G, Najjar S, Daher R. False-negative primary neonatal thyroid screening: the need for clinical vigilance and secondary screening. J Med Screen 2003; 10 (1): 2-4.
6.- Mengreli C, Kanaka-Gantenbein C, Girginoudis P. Screening for congenital hypothyroidism: the significance of threshold limit in false-negative results. J Clin Endocrinol Metab 2010; 95 (9): 4283-90.
7.- Geelhoed EA, Lewis B, Hounsome D, O’Leary P. Economic evaluation of neonatal screening for phenylketo-nuria and congenital hypothyroidism. J Paediatr Child Health 2005; 41 (11): 575-9.
8.- Carroll AE, Downs SM. Comprehensive cost-utility analysis of newborn screening strategies. Pediatrics 2006; 117 (5 Pt 2): S287-95.
9.- Grosse SD, Van-Vliet G. Prevention of intellectual disability through screening for congenital hypothyroidism: how much and at what level?. Arch Dis Child 2011; 96 (4): 374-9.
10.- Al-Taji E, Biebermann H, Limanova Z. Screening for mutations in transcription factors in a Czech cohort of 170 patients with congenital and early-onset hypo-thyroidism: identification of a novel PAX8 mutation in dominantly inherited early-onset non-autoimmune hypothyroidism. Eur J Endocrinol 2007; 156 (5): 521-9.
11.- La-Franchi SH. Approach to the diagnosis and treatment of neonatal hypothyroidism. J Clin Endocrinol Metab 2011; 96 (10): 2959-67.
12.- Rastogi MV, La-Franchi SH. Congenital hypothyroidism. Orphanet J Rare Dis 2010; 5: 17.
13.- Jacobsen BB, Brandt NJ. Congenital hypothyroidism in Denmark. Arch Dis Child 1981; 56 (2): 134-6.
14.- Tarim OF, Yordam N. Congenital hypothyroidism in Turkey: a retrospective evaluation of 1000 cases. Turk J Pediatr 1992; 34 (4): 197-202.
15.- La-Franchi SH. Hypothyroidism. Pediatr Clin North Am 1979; 26 (1): 33-51.
16.- Grasberger H, Refetoff S. Genetic causes of congenital hypothyroidism due to dyshormonogenesis. Curr Opin Pediatr 2011; 23 (4): 421-8.
17.- Peter F, Muzsnai A. Congenital disorders of the thyroid: hypo/hyper. Pediatr Clin North Am 2011; 58 (5): 1099-115.
18.- Grant DB, Smith I, Fuggle PW, Tokar S, Chapple J. Congenital hypothyroidism detected by neonatal screening: relationship between biochemical severity and early clinical features. Arch Dis Child 1992; 67 (1): 87-90.
19.- Olivieri A, Stazi MA, Mastroiacovo P. A po- pulation-based study on the frequency of additional congenital malformations in infants with congenital hypothyroidism: data from the Italian Registry for Congenital Hypothyroidism (1991-1998). J Clin Endocrinol Metab 2002; 87 (2): 557-62.
20.- American Academy of P, Rose SR, Section on E, Committee on Genetics ATA, Brown RS, Public Health Committee LWPES, et al: Update of newborn screening and therapy for congenital hypothyroidism. Pediatrics 2006; 117 (6): 2290-303.
21.- Ohnishi H, Sato H, Noda H, Inomata H, Sasaki N. Color Doppler ultrasonography: diagnosis of ectopic thyroid gland in patients with congenital hypothyroidism caused by thyroid dysgenesis. J Clin Endocrinol Metab 2003; 88 (11): 5145-9.
22.- Brown RS, Bellisario RL, Botero D. Incidence of transient congenital hypothyroidism due to maternal thyrotropin receptor-blocking antibodies in over one million babies. J Clin Endocrinol Metab 1996; 81 (3): 1147-51.
23.- Glinoer D. Pregnancy and iodine. Thyroid 2001; 11 (5): 471-81.
24.- Becerra C. Hipotiroidismo congénito y fenilcetonuria en el niño. RevChil Pediatr 2008; 79: 96-102.
25.- Selva KA, Harper A, Downs A, Blasco PA, Lafranchi SH. Neurodevelopmental outcomes in congenital hypo-thyroidism: comparison of initial T4 dose and time to reach target T4 and TSH. J Pediatr 2005;147 (6): 77-580.
26.- Ng SM, Anand D, Weindling AM. High versus low dose of initial thyroid hormone replacement for congenital hypothyroidism. Cochrane Database Syst Rev 2009 (1): CD006972.
Los autores declaran no tener conflictos de interés, ni haber recibido aportes financieros de ningún tipo. La Dra. Francisca Grob ha recibido una beca de la Sociedad Chilena de Endocrinología y Diabetes (SOCHED) y de la Sociedad Latinoamericana de Endocrinología Pediátrica (SLEP) para realizar una estadía de endocrinología molecular en el Centre Hospitalier Universitaire Sainte Justine, en Montreal, Canadá.


Más notas de la edición 71
Lee desde Issuu nuestra última edición publicada en Diciembre 2025, Edición número 172
Notas relacionadas a Hipotiroidismo congénito: un diagnóstico que no...


