

Hemoglobinuria paroxística nocturna: Una causa infrecuente y tratable de lesión renal aguda. Caso clínico
La hemoglobinuria paroxística nocturna (HPN) es un trastorno clonal poco frecuente de las células hematopoyéticas (CH), caracterizado por anemia hemolítica, insuficiencia medular en grado variable y trombosis1.
Raimundo Contreras1 2
Gonzalo P Méndez3
José Mujica4
Carlos Padilla2 5
María E. Selamé5 6
Alondra Frías5 6 *
1Becado de Medicina Interna, Universidad Católica del Maule. Talca, Chile. 2Servicio de Medicina Interna, Hospital Regional de Talca. Talca, Chile. 3Sección de Anatomía Patológica, Laboratorio Inmunocel. Santiago, Chile. 4Servicio de Hematología, Hospital Regional de Talca. Talca, Chile. 5Facultad de Medicina, Universidad Católica del Maule. Talca, Chile. 6Servicio de Nefrología, Hospital Regional de Talca. Talca, Chile.
*Correspondencia: Alondra Frías / afrias@ hospitaldetalca.cl 1 Norte #1990, Talca, Chile. Servicio de Nefrología.
Resumen
La detección precoz de lesión renal aguda es fundamental para prevenir la progresión a enfermedad renal crónica. Presentamos el caso de un hombre de 19 años, con múltiples consultas en urgencias por cuadro de fatigabilidad, dolor abdominal y orina oscura intermitente. Al ingreso destacaba pancitopenia con elementos sugerentes de hemólisis y lesión renal aguda. Se sospechó púpura trombocitopénico trombótico por lo que fue tratado con dexametasona y plasmaféresis. El estudio complementario descartó esta sospecha, siendo suspendida la plasmaféresis. La biopsia renal reveló lesión renal aguda compatible con hemoglobinuria paroxística nocturna (HPN) y la biopsia de médula ósea excluyó infiltración medular. Se confirmó el diagnóstico de HPN en citometría de flujo. Fue tratado con prednisona, anticoagulación, suplementación de hierro y ácido fólico. Evolucionó con respuesta clínica y de laboratorio favorable.
Palabras clave: Hemólisis, Hemoglobinuria Paroxística Nocturna, Lesión Renal Aguda,Plasmaféresis.
Abstract: Paroxysmal Nocturnal Hemoglobinuria: A Rare but Treatable Cause of Acute Kidney Injury. Case Report
Early recognition of acute kidney injury is essential to prevent progression to chronic kidney disease. We present the case of a 19-year-old man with multiple emergency department visits for fatigue, abdominal pain, and intermittent dark urine. Upon admission, he had pancytopenia with elements suggestive of hemolysis and acute kidney injury. Thrombotic thrombocytopenic purpura was suspected, so he was treated with dexamethasone and plasmapheresis. The complementary study ruled out this suspicion, and plasmapheresis was suspended. The renal histology showed an acute kidney injury consistent with paroxysmal nocturnal hemoglobinuria (PNH), and bone marrow biopsy excluded marrow infiltration. The diagnosis of PNH was confirmed by flow cytometry. He was treated with prednisone, anticoagulation, iron, and folic acid supplementation. He progressed with a favorable clinical and laboratory response.
Keywords: Acute Kidney Injury, Hemolysis, Paroxysmal Nocturnal Hemoglobinuria, Plasmapheresis.
La hemoglobinuria paroxística nocturna (HPN) es un trastorno clonal poco frecuente de las células hematopoyéticas (CH), caracterizado por anemia hemolítica, insuficiencia medular en grado variable y trombosis1.
La HPN tiene una prevalencia de 0,5 a 1 caso por 100.000 habitantes, alcanzando un peak entre los 30 a 40 años2. En Chile no existen datos de prevalencia3.
Esta enfermedad es causada por una mutación en el gen fosfatidilinositol glicano de clase A (PIG-A), encargado de la síntesis de glicofosfatidilinositol (GPI) que permite el anclaje de proteínas moduladoras del complemento (CD55 y CD59) a las membranas de las CH1,4. La ausencia de anclaje de estas proteínas determina que exista hemólisis mediada por complemento.
La HPN también puede manifestarse clínicamente con cefalea, dolor abdominal o lumbar, orina oscura, ictericia, eventos trombóticos en diferentes sistemas y complicaciones nefrológicas1,4. La HPN es una causa infrecuente de lesión renal aguda (LRA), la que puede estar determinada por múltiples mecanismos de daño. A continuación, presentamos el reporte de un paciente adulto que presentó LRA en el contexto de una crisis de HPN.
Caso clínico
Hombre de 19 años, con múltiples consultas por cuadro de tres meses de evolución caracterizado por fatigabilidad, dolor abdominal en hipocondrio derecho, de carácter sordo, moderada intensidad e intermitente, asociado a cefalea opresiva holocránea y orina oscura.
Consultó al servicio de urgencia donde se constató presión arterial 117/65 mmHg, frecuencia cardíaca 62 latidos por minutos, frecuencia respiratoria 12 respiraciones por minuto, temperatura 36,4°C, peso 60 kg, débito urinario 0,6 ml/kg/hora en las últimas 6 horas, ictericia leve y abdomen doloroso difuso, sin signos de irritación peritoneal ni hipovolemia.
Se hospitalizó para estudio y tratamiento por sospecha de púpura trombocitopénico trombótico (PTT, PLASMIC score 4 puntos). Recibió 2.000 mL de solución salina al 0,9% las primeras 24 horas con aumento del débito urinario a 1,3 mL/kg/hora, plasmaféresis (PLEX) y dexametasona 20 mg/día intravenosa (IV).
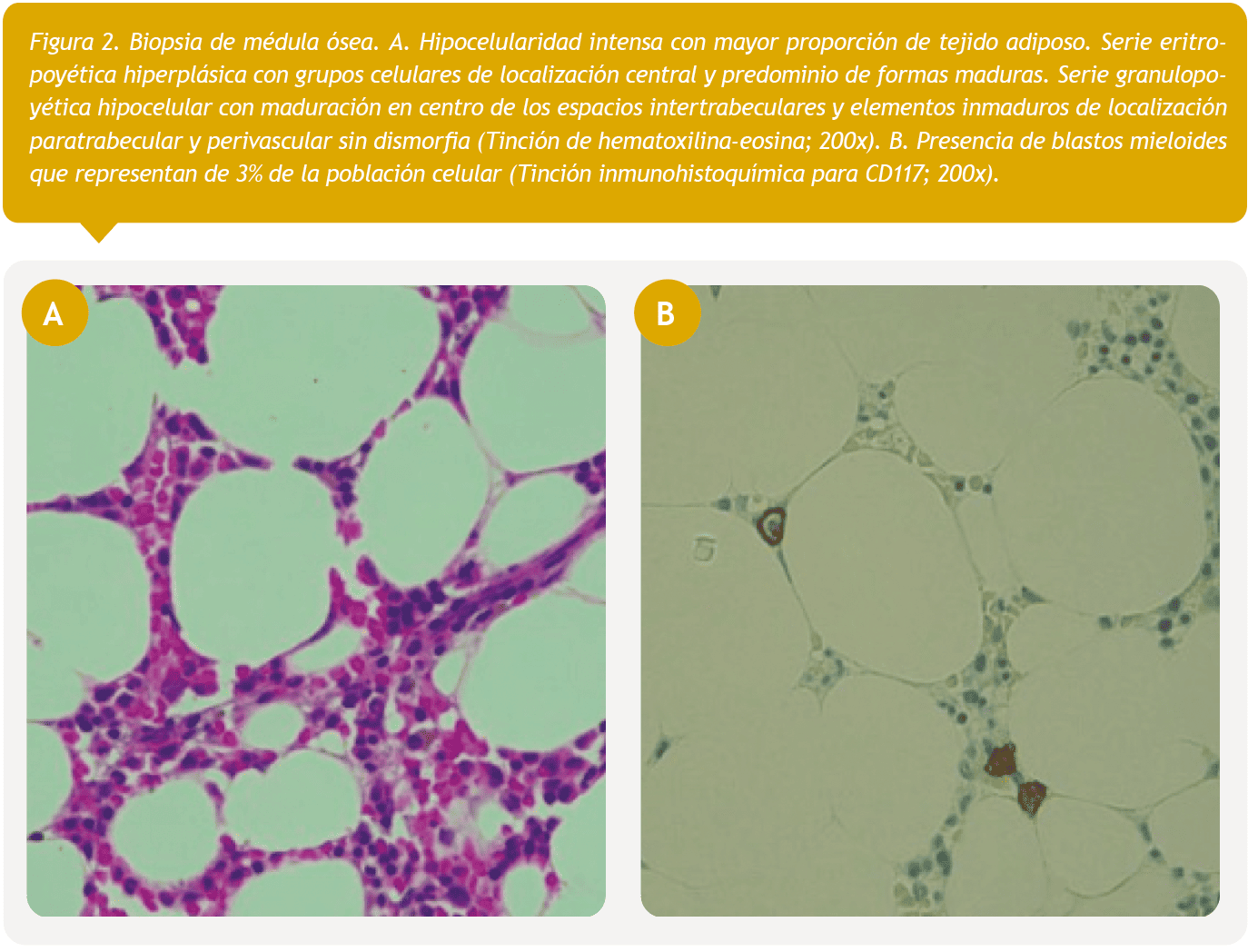
El estudio inicial (Tabla 1) no identificó esquistocitos en el frotis, el test de Coombs fue negativo y no estaba disponible medición de haptoglobina. Se pesquisó síndrome respiratorio agudo grave por coronavirus de tipo 2 (SARS-CoV-2) asintomático.
El estudio restante fue negativo o normal para: ADAMTS 13 (actividad 140%, referencia >40%, actividad metaloproteinasa de FvW no disponible), vitamina B12 (265 pg/mL, referencia 197–771 pg/mL), cocaína, Elisa VIH, HBsAg, anticuerpos (AC) anti VHC, IgM e IgG para Epstein Barr y Citomegalovirus, C3 (92 mg/dL, referencia 90-180 mg/dL), C4 (32 mg/dL, referencia 10-40 mg/dL), AC antinucleares, AC anti DNAds, anticoagulante lúpico, AC anticardiolipinas, AC anti β2-glicoproteína, cadenas livianas en sangre, electroforesis e inmunofijación en sangre y orina.
Tomografía computarizada de tórax, abdomen y pelvis con contraste descartó trombosis, neoplasia, visceromegalias y adenopatías.

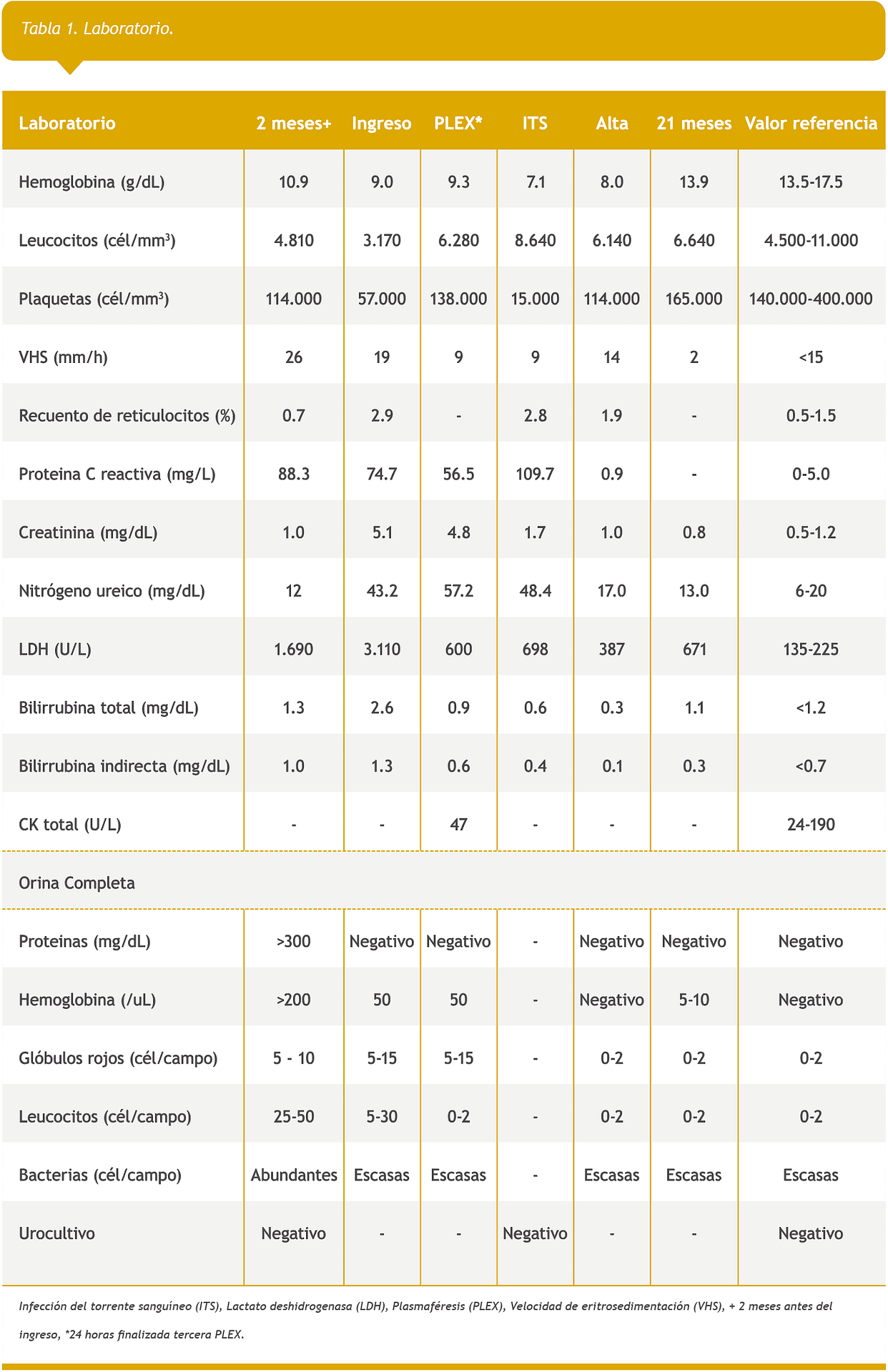
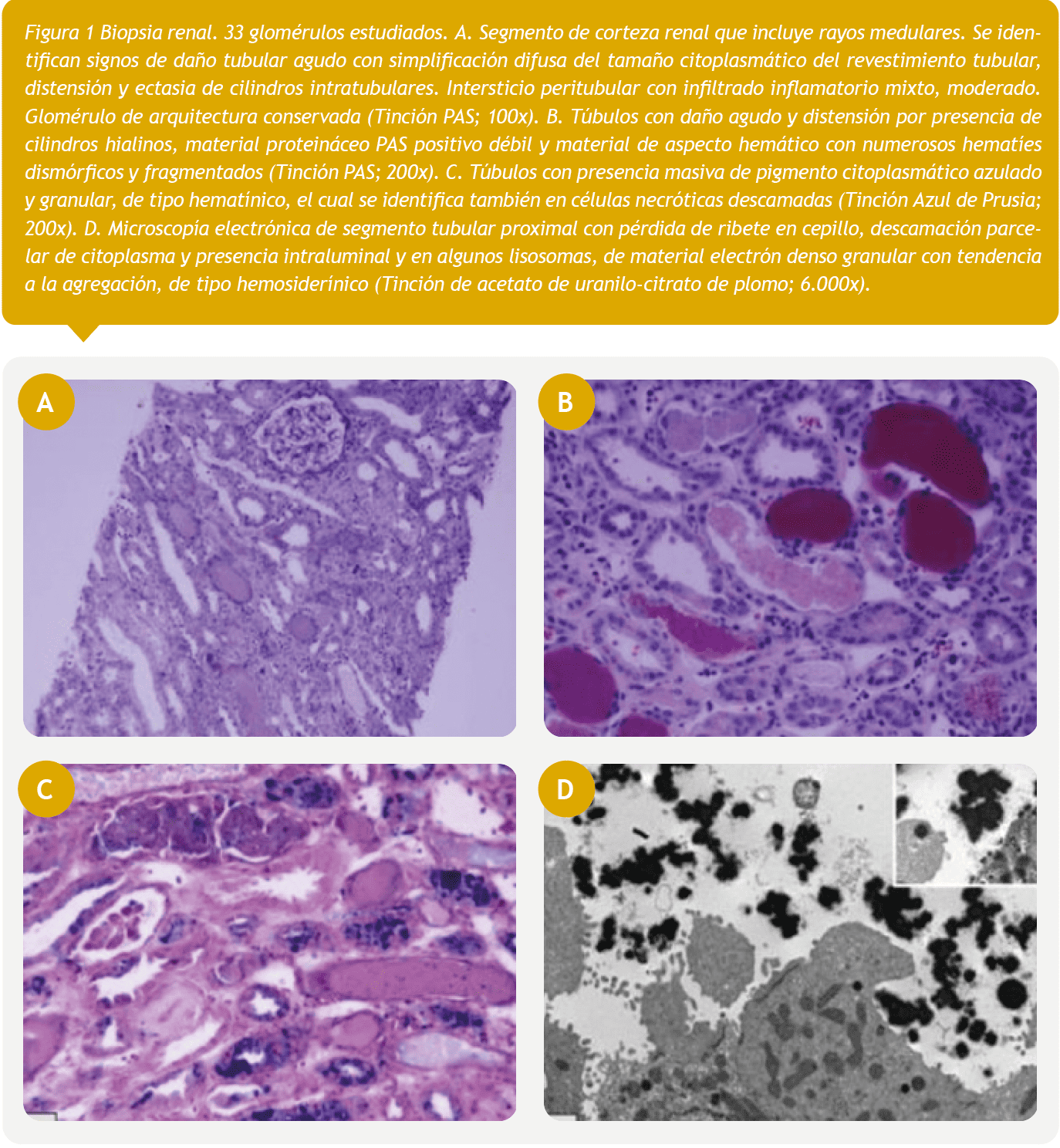
Se realizó biopsia renal que evidenció extensos signos de daño tubular agudo asociado a abundante pigmento hemosiderínico (Figura 1). El mielograma informó plasmocitosis de 4%, sin compromiso de otras series y la biopsia de médula ósea concluyó hipocelularidad severa en ausencia de infiltración medular (Figura 2).
La citometría de flujo e inmunofenotipo para HPN fueron compatibles e identificaron clon en eritrocitos (56,12%) y granulocitos (84,31%). No se encontraba disponible estudio con proaerolisina marcada con fluoresceína.
El estudio complementario descartó PTT siendo suspendida la PLEX al tercer día. Dexametasona 20 mg/día IV fue administrada durante seis días, luego prednisona 60 mg/día oral. Evolucionó con ascenso del recuento de plaquetas (Tabla 1).
El día 18 de hospitalización, cursó con infección del torrente sanguíneo (ITS) asociada a catéter por S. aureus multisensible tratada 14 días con Cefazolina 2 g cada 8 horas IV. En este periodo presentó anemización y trombocitopenia (Tabla 1), sin requerimiento de transfusión de hemoderivados.
Tras este episodio se inició anticoagulación con rivaroxabán 15 mg cada 12 horas oral, ajustó prednisona a 40 mg/día oral, suplementó hierro oral y ácido fólico e inmunizó contra hepatitis B, neumococo, meningococo e influenza.

Evolucionó con recuperación completa de pancitopenia y LRA. En la actualidad se mantiene asintomático, recibiendo tratamiento anticoagulante oral, sin corticoides y en espera de iniciar inhibidor del complemento solicitado desde la hospitalización.
Discusión
La HPN es una alteración clonal adquirida de las CH, sin predisposición por sexo y de mayor frecuencia en la tercera a cuarta década de la vida5. Nuestro caso tiene una presentación más temprana.
Se genera por una mutación en el cromosoma X en el gen PIG-A, que altera la síntesis de GPI, evitando el anclaje de las glicoproteínas CD55 y CD59 a la membrana de las CH1,6. Es así como, se produce una activación descontrolada del complemento por reducción o ausencia de estas glicoproteínas causando hemólisis crónica, activación plaquetaria y trombosis1,4.
En ausencia de CD55 se activa la convertasa de C3 e indirectamente se constituye el complejo de ataque de membrana (CAM), esto determina hemólisis intra y extravascular. La deficiencia de CD59 activa el CAM que produce hemólisis intravascular6.

Se recomienda descartar HPN en escenarios como el de este reporte, en que están presentes pancitopenia, anemia hemolítica con test de Coombs negativo, dolor abdominal sin etiología y hemoglobinuria4.
Para diagnosticar HPN es necesario realizar un amplio diagnóstico diferencial y excluir otras causas de microangiopatía trombótica y de daño endotelial: lupus eritematoso sistémico, síndrome antifosfolípido catastrófico, crisis renal esclerodérmica, neoplasias hematológicas, infecciones virales crónicas, cocaína y fármacos (Gemcitabina, Bevacizumab e inhibidores de calcineurina)7.
En el caso presentado, la indicación de PLEX se pudo haber reconsiderando debido al bajo riesgo para PTT (PLASMIC score 4 puntos) y haber esperado el resultado de la actividad de ADAMTS 13. El score PLASMIC tiene una alta sensibilidad como un alto valor predictivo negativo y es recomendado para escenarios en que no se dispone en 72 horas de este examen21.
El estudio de elección para confirmar HPN es la citometría de flujo en sangre periférica, examen de alto costo6,8. En este reporte, la citometría de flujo fue concordante con HPN, identificando clones en eritrocitos y granulocitos.
Aun cuando la HPN es una causa inhabitual de LRA, tanto la enfermedad renal crónica (ERC) como la LRA son complicaciones frecuentes de HPN9. En la literatura han sido caracterizadas en forma variable según la serie estudiada, siendo descritas desde 3 a 14,6%10 para LRA y de 8,6 a 21% para ERC11. Son múltiples los mecanismos implicados en LRA: daño tubular, obstrucción tubular, hipovolemia, isquemia cortical, trombosis bilateral de las venas renales, oclusión de la arteria renal en forma menos frecuente y depósito glomerular de fibrina5,9,12. El daño tubular es secundario a la metabolización del grupo hemo a hemosiderina en el túbulo proximal, que determina depleción de óxido nítrico, vasoconstricción intrarrenal e infartos corticales5,9. Otros procesos involucrados son: estrés oxidativo, obstrucción tubular por cilindros de hemoglobina y ácido úrico5,9, peroxidación de lípidos y apoptosis13,14.

La resonancia nuclear magnética es útil para localizar microinfartos corticales asintomáticos. Es capaz de detectar hemosiderina cortical, identificada como diferenciación inversa de la corteza y médula en T1, y pérdida sustancial de la intensidad de la señal cortical en T1 y T2 14.
Los mecanismos involucrados en ERC son hemólisis crónica que genera exposición repetida del túbulo proximal a hemosiderina, síndrome de Fanconi y necrosis cortical por trombosis microvascular recurrente. Esto determina a largo plazo atrofia tubular y fibrosis intersticial9,15.
La biopsia renal tiene un gran valor en el diagnóstico y pronóstico, permite confirmar daño tubular agudo caracterizado por depósito intenso de hemosiderina en el túbulo proximal, a través de tinción con azul de Prusia (Figura 1C en el caso presentado). Otros elementos son: depósito de cristales de ácido úrico y necrosis papilar16. La presencia de fibrosis intersticial y glomeruloesclerosis secundaria son marcadores de ERC16. En este reporte la presencia de fibrosis intersticial era menor al 10%.
El tratamiento de la HPN considera el uso de inhibidores del complemento (IC) y en algunos casos trasplante de médula ósea alogénica. Otras medidas complementarias son: corticoides sistémicos, transfusiones y anticoagulación 1,17.
Eculizumab y Ravulizumab, son anticuerpos monoclonales humanizados dirigidos contra C5, que impiden la activación crónica y descontrolada del complemento. Ambos han cambiado el curso natural de la enfermedad al aumentar la sobrevida, permitiendo que sea actualmente similar a la de la población general17.
Estos IC han mejorado la calidad de vida y tienen un adecuado perfil de seguridad, reduciendo la hemólisis y el número de eventos trombóticos en forma significativa4,17. Ravulizumab es superior a Eculizumab debido a una vida media más larga y a una menor probabilidad de recidiva de hemólisis respecto a Eculizumab (4% y 11% respectivamente)4,20. En nuestro caso el uso de IC está pendiente debido a su alto costo.
Otra alternativa de tratamiento para la HPN es Pegcetacoplan, un pentadecapéptido pegilado que inhibe C3 y previene la hemólisis extravascular. El estudio PEGASUS comparó Pegcetacoplan versus Eculizumab y encontró mayores niveles de hemoglobina luego de 16 semanas de tratamiento10.
En marzo del 2024, fue publicado un estudio de fase 3 que utilizó Iptacopan oral como monoterapia para tratar a pacientes con anemia persistente tratados con anterioridad con y sin inhibidor de C5. En ambos grupos aumentó la hemoglobina hasta en 2 gr/dL y alcanzó niveles de 12 gr/dL 18.
El trasplante de médula ósea es considerado como una terapia de segunda línea, por el menor beneficio en comparación con los IC y su alta morbimortalidad1,4.
Los corticoides son utilizados para la prevención y tratamiento de las crisis de hemólisis. Sin embargo, el mecanismo de acción aún no está claro, probablemente inhiben la activación del complemento17. En este reporte se administraron corticoides durante la crisis de HPN, logrando normalización de hemoglobina y plaquetas posterior al alta.
Factores que pueden gatillar hemólisis son infecciones8,13,14, cirugías, trauma y embarazo13. En nuestro caso, proponemos como explicación a los exámenes presentados en la tabla 1 dos meses antes de la hospitalización una infección urinaria baja que pudo haber gatillado hemólisis y durante la infección por SARS-CoV-2. Las proteínas spikes (PS) del SARS-CoV-2 pueden activar todas las vías del complemento. La vía alterna se activa por unión de las PS al heparán sulfato que compiten con el factor H13,19. Crisis hemóliticas asociadas a infección por SARS-CoV-2 han sido reportadas por el grupo de Yan Hui19. Por estos motivos, es necesario prevenir infecciones, ya que todas estas condiciones son amplificadoras del complemento.
Finalmente, es fundamental el estudio etiológico precoz y exhaustivo de la LRA, destacando el rol de la biopsia renal, pues en este reporte hizo posible identificar una causa infrecuente que tiene un tratamiento específico y evitar la progresión a ERC.
Concluimos que la HPN es una patología que requiere una alta sospecha y, como fue descrito es necesario realizar un detallado diagnóstico diferencial. En la actualidad, los IC permiten un mejor tratamiento de la enfermedad y una mayor sobrevida.
Financiamiento: Este trabajo no contó con apoyo financiero de ningún tipo.
Agradecimientos: A los doctores Lucas Videla y Julio Pérez, quienes facilitaron imágenes de médula ósea para este reporte.
Referencias Bibliográficas
1 Brodsky R. Paroxysmal nocturnal hemoglobinuria. Blood. 2014; 124(18): 2804-2811.
2 Jalbert J, Chaudhari U, Zhang H, Weyne J, Shammo J. Epidemiology of PNH and Real-World Treatment Patterns Following an Incident PNH Diagnosis in the US. Blood. 2019; 134(1): 3407.
3 Arruda M, Rodrigues C, Yamamoto M, Figueiredo M. Paroxysmal nocturnal hemoglobinuria: From physiopathology to treatment. Revista da Associacao Medica Brasileira. 2010; 56(2): 214-221.
4 Cançado R, Araújo A, Sandes A, Arrais C, Castro C, Figueiredo M. Consensus statement for diagnosis and treatment of paroxysmal nocturnal haemoglobinuria. Hematology Transfusion and Cell Therapy. 2021; 43(3): 341-348.
5 Ram R, Adiraju K, Gudithi S, Dakshinamurty K. Renal Manifestations in Paroxysmal Nocturnal Hemoglobinuria. Indian Journal of Nephrology. 2017; 27(4): 289-293.
6 Szlendak U, Budziszewska B, Spychalska J, Drozd-Sokolowska J, Patkowska E, Nowak J. Paroxysmal nocturnal hemoglobinuria: Advances in the understanding of pathophysiology, diagnosis, and treatment. Polish archives of Internal Medicine. 2022; 132(6): 16271.
7 Mondragon A, Rojas J, Jimenez C, Umana H. Hemoglobinuria Paroxística Nocturna: reporte de un caso. Revista Investigaciones Andina. 2013; 15(26): 716-723.
8 Parker C. Update on the diagnosis and management of paroxysmal nocturnal hemoglobinuria. Hematology American Society of Hematology. Education Program. 2016; 1: 208-216.
9 Kokoris S, Gavriilaki E, Miari A, Travlou A, Kyriakou E, Anagnostopoulos A. Renal involvement in paroxysmal nocturnal hemoglobinuria: An update on clinical features, pathophysiology and treatment. Hematology. 2008; 23(8): 558-566.
10 Hillmen P, Elebute M, Kelly R, Urbano-Ispizua A, Hill A, Russell P. Long-term effect of the complement inhibitor eculizumab on kidney function in patients with paroxysmal nocturnal hemoglobinuria. American Journal of Hematology. 2010; 85(8): 553-559.
11 Jang JH, Kim JS, Yoon SS, Lee JH, Kim YK, Jo DY. Predictive Factors of Mortality in Population of Patients with Paroxysmal Nocturnal Hemoglobinuria (PNH): Results from a Korean PNH Registry. Journal of Korean Medicine Science. 2016; 31(2): 214-221.
12 Madkaikar M, Gupta M, Jijina F, Ghosh K. Paroxysmal nocturnal haemoglobinuria: Diagnostic tests, advantages, & limitations. European Journal of Haematology. 2009; 83(6): 503-511.
13 Yu J, Yuan X, Chen H, Chaturvedi S, Braunstein E, Brodsky R. Direct activation of the alternative complement pathway by SARS-CoV-2 spike proteins is blocked by factor D inhibition. Blood. 2020; 136(18): 2080-2089.
14 Kim Y, Yang J, Choi J, Kim B, Yu J, Kim Y. Hemolytic Crisis and Acute Kidney Injury in patient with Paroxysmal Nocturnal Hemoglobinuria in Korea-Case Report and Review of Literature. The Korean Journal of Nephrology. 2009; 28: 236-242.
15 Brocklebank V, Wood K, Kavanagh D. Thrombotic Microangiopathy and the Kidney. CJASN. 2018; 13(2): 300-317.
16 Puri V, Gandhi A, Sharma S. Renal Biopsy in Paroxysmal Nocturnal Hemoglobinuria: An Insight into the Spectrum of Morphologic Changes. Indian Journal of Nephrology. 2017; 27(4): 284-288.
17 Guías Prácticas Clínicas para Diagnóstico y Tratamiento de la Hemoglobinuria Paroxística Nocturna. Sociedad Chilena de Hematología. Ministerio de Salud, Chile. 2020.
18 Peffault R, Röth A, Kulasekararaj A, Han B, Scheinberg P, Maciejewski J. Oral Iptacopan monotherapy in paroxysmal nocturnal hemoglobinuria. New England Journal of Medicine. 2024; 390: 994-1008.
19 Yang H, Chai X, Gong Y, Zhang X, Wang L, Zhou X. Severe hemolytic exacerbations of Chinese PNH patientsinfected SARS-CoV-2 Omicron. Immunity, Inflamation and Disease. 2023; 11(8): e966.
20 Brodsky R, Peffault R, Rottinghaus S, Röth A, Risitano A, Weitz I. Characterization of breakthrough hemolysis events observed in the phase 3 randomized studies of ravulizumab versus eculizumab in adults with paroxysmal nocturnal hemoglobinuria. Haematologica. 2021; 106(1), 230-237.
21 Paydary K, Banwell E, Tong J, Chen Y, Cuker A. Diagnostic accuracy of the PLASMIC score in patients with suspected thrombotic thrombocytopenic purpura: A systematic review and meta-analysis. Transfusion. 2020; 60(9): 2047-2057.
Recibido: 19 de Enero de 2024; Aprobado: 28 de Junio de 2024
*Correspondencia: Alondra Frías / afrias@hospitaldetalca.cl 1 Norte #1990, Talca, Chile. Servicio de Nefrología.
Los autores declaran no presentar conflicto de interés.
Creative Commons License, Imagen This is an Open Access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.


Más notas de la edición 165
Lee nuestra última edición publicada en Julio 2026, Edición número 179

Notas relacionadas a Hemoglobinuria paroxística nocturna: Una causa...

