
Glucogenosis Tipo I: Enfermedad de Von Gierke
Asociación Española de Enfermos de Glucogenosis
Javier Fernandez Salido, Alberto Molares Vila, Jesus Sueiro Justel, Juan J. Albero Samper, Benjamín Anton, Antonio M. Banon Hernandez, Laura Castells Molines, Jose Luis Ceide Arias, Leonor Fernandez Marcos, Maria Jose Santos García
E-mail: amhernan@ual.esE-mail; amolares@gmail.com
Introducción
Las glucogenosis pertenecen al grupo de enfermedades raras. Esto ha determinado que muchas personas afectadas no cuenten en su zona de residencia con especialistas cualificados y que la información recibida en muchos casos sea escasa, incompleta y, a veces, errónea. Uno de los grandes problemas con los que se encuentran los afectados por glucogenosis es la falta de información.
Con esta guía se pretende, por una parte, presentar a los afectados y familiares un resumen de la información publicada sobre la glucogenosis tipo I, abarcando los diferentes aspectos de esta enfermedad y, por otra, despertar entre la comunidad médica el interés por esta patología. El objetivo último es que se aúnen esfuerzos, aportando aquellos sus experiencias en el día a día y estos sus conocimientos e investigaciones, para alcanzar un futuro cada vez mejor para todos los afectados.Otras asociaciones como la francesa y la inglesa han conseguido en estos últimos años implicar a reconocidos investigadores, dietistas y médicos que han contribuido sobremanera a mejorar la información disponible sobre los distintos tipos de glucogenosis y, sobre todo, la calidad de vida de las personas afectadas por las mismas. Este es también el objetivo de nuestra asociación.
¿Qué es la glucogenosis tipo I?
La glucogenosis tipo I (GSD-I) es una enfermedad metabólica, rara y hereditaria, provocada por deficiencias en el sistema de la Glucosa-6-Fosfatasa (G-6-Fosfatasa) [1-2]. Este sistema se compone de 4 proteínas: por una parte, la enzima catalizadora glucosa-6-fosfatasa, que transforma la glucosa-6-fosfato proveniente del glucógeno hepático y de la gluconeogénesis en glucosa (la deficiencia de esta enzima provoca la GSD tipo Ia); y por otra, las enzimas transportadoras de la glucosa-6-fosfato (su deficiencia provoca la GSD tipo Ib), del fosfato inorgánico (su deficiencia se cree que provoca la GSD tipo Ic) y de la glucosa libre (su deficiencia se cree que provoca la GSD tipo Id). La enfermedad fue diagnosticada por primera vez en 1928 por Van Greveld, y estudiada histológicamente por Von Gierke en 1929 [3].
Sinónimos
Glycogen Storage Disease Type I (GSD-I)
Enfermedad de Von GierkeGlucogenosis Hepatorrenal
Deficiencia de Glucosa-6-FosfatasaEntrada nº 232200 en McKusick´s catalogue: Mendelian Inheritance in Man (OMIM) [4].
La enfermedad de Von Gierke o Glucogenosis tipo I, puede incluirse en cualquiera de las siguientes categorías:
- Glucogenosis
- Errores innatos del metabolismo
- Enfermedades de depósito de glucógeno
- Enfermedades genéticas
- Enfermedades raras
Subtipos Clínicos
Dentro de la GSD-I los dos subtipos de mayor incidencia son el Ia y el Ib. Se estima que el subtipo Ia es el más común, y está explicado por un déficit de actividad de la enzima G-6-Fosfatasa en el ámbito hepático y renal [5-7]. Por otra parte, también es relativamente frecuente encontrar pacientes con características clínicas prácticamente indistinguibles de la GSD-Ia, pero con niveles normales de actividad enzimática in vitro de G-6-Fosfatasa. Se ha demostrado una deficiencia del transporte de la glucosa-6-fosfato en esta variedad de la enfermedad, clasificada como GSD-Ib o pseudotipo I. Más recientemente, se han podido describir otros dos tipos más raros (Ic y Id), también caracterizados por deficiencias en las enzimas transportadoras en el sistema de la G-6-Fosfatasa, pero que aún no son totalmente distinguibles del subtipo Ib, por lo que todavía existen controversias en cuanto a su categorización.
Las diferencias clínicas entre el tipo Ia y el Ib no son significativas, con la particularidad de que los afectados por el tipo Ib presentan, además, infecciones bacterianas recurrentes y neutropenia (niveles anormalmente bajos de neutrófilos). Estos últimos, también pueden desarrollar inflamación crónica del intestino.
Recientemente se ha sugerido que, en el caso del tipo Ia, la deficiencia en la homeostasis de la glucosa puede comprometer al sistema inmune, dando lugar a un aumento en la concentración de los glóbulos blancos y en la producción de citoquinas [8]. También se ha demostrado que pacientes con glucogenosis tipo Ia exhiben un elevado número de neutrófilos periféricos y de interleucina-8 (IL-8) sérica en comparación con controles sanos. Así, se encontraron concentraciones elevadas de IL-8 (proteína inflamatoria) en pacientes afectados por la tipo Ia con adenomas hepáticos [9].
Incidencia
Se estima que la incidencia es del orden de uno cada 100.000 nacimientos. La enfermedad de Von Gierke se transmite de forma autosómica recesiva. La herencia de las enfermedades genéticas se describe tanto por el tipo de cromosoma en que se encuentra el gen anormal (autosómico o cromosoma sexual), como por el hecho de que el mismo gen sea dominante o recesivo. Si es dominante, el gen anormal de uno de los padres es suficiente para provocar la enfermedad y, si es recesivo, es necesario que ambos genes sean anormales para que se produzca la enfermedad. Por tanto, la GSD-I está presente tanto en hombres como en mujeres y es necesario que ambos padres transmitan el gen mutado para que esta enfermedad se manifieste.
Estadísticamente, si ambos padres son portadores del gen mutado, cada uno de sus hijos tiene el 25% de probabilidad de heredar la enfermedad, el 50% de ser portador sin desarrollar la enfermedad, y el 25% de no ser portador.
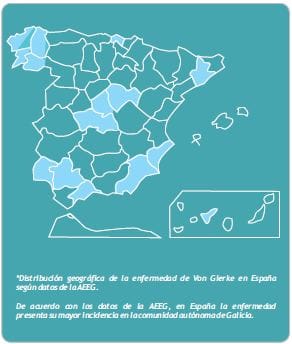
Mortalidad
Las principales causas de muerte son convulsiones hipoglucémicas y/o acidosis grave [10]. En la GSD-Ib, las infecciones pueden ser una causa probable de muerte [11]. Es posible, por otra parte, que se produzca hipoglucemia profunda sin síntomas clínicos. Este fenómeno se explica mediante la elevación de la concentración de lactato en sangre, que sustituye a la glucosa como fuente de energía para el cerebro.
La mortalidad, frecuente en otras épocas, se ha tornado ahora en rara si el control metabólico es el adecuado [12-13].
Causas de la Glucogenosis Tipo I
En esta forma de glucogenosis, el defecto básico es que el paciente no puede convertir la glucosa-6-fosfato en glucosa libre (sustancia de la que el organismo obtiene energía). El problema inmediato es la baja cantidad de azúcar en la sangre; como consecuencia de ello, algunos pacientes, sobre todo niños, tienen un alto riesgo de sufrir profundas hipoglucemias. Aunque el error metabólico está centrado en el hígado, también puede existir deficiencia de la enzima en los riñones e intestino delgado.
En las personas sanas, el hígado almacena glucosa en forma de glucógeno (usualmente hasta 5 gr. de glucógeno cada 100 gr. de tejido hepático), de manera que, cuando el azúcar en sangre cae, este glucógeno se convierte en glucosa libre y conserva el nivel de azúcar normal en sangre (normoglucemia).

Como los pacientes con GSD-I pueden almacenar glucosa como glucógeno pero no pueden liberarlo normalmente, con el tiempo se acumulan grandes cantidades de glucógeno en el hígado. Ciertas hormonas, particular-mente el glucagón, se incrementan en el cuerpo en un vano intento por parte del organismo de hacer crecer el nivel de azúcar en la sangre. También aumentan considerablemente el ácido láctico y las grasas en la sangre. Las grasas se movilizan y se almacenan en el hígado (generando el efecto de hígado graso) junto con el glucógeno, lo que conduce a un agrandamiento del hígado (hepatomegalia). Por lo demás, el hígado funciona con normalidad.
Síntomas de la Enfermedad de Von Gierke
La enfermedad puede manifestarse en los primeros meses de vida, o bien, en los casos menos graves, hacia finales del primer año. Sin embargo, en una comunicación reciente [54] se presenta el caso de un paciente diagnosticado a los 42 años por la presencia de adenomas hepáticos, con síntomas muy leves en la infancia, lo que se explicaría por una actividad enzimática residual. Los recién nacidos pueden presentar hepatomegalia, distrés respiratorio, lactacidosis y convulsiones hipoglucémicas.
En la Niñez:
- Hipoglucemia: niveles de azúcar en la sangre muy bajos.
- Ausencia de respuesta a la prueba de glucagón o adrenalina: se incrementa el ácido láctico en sangre en lugar de los niveles de azúcar.
- Hepatomegalia: agrandamiento del hígado.
- Aspecto de “muñeca”: mejillas hinchadas, extremidades y tórax delgados y un vientre protuberante.
- Intolerancia al ayuno, necesidad de alimentaciones frecuentes.
- Niveles altos de ácido láctico, colesterol y grasas en sangre (principalmente triglicéridos).
- Retraso en el crecimiento lineal y del desarrollo motor.
- Sangrados frecuentes y hematomas por deficiencias plaquetarias.
- Neutropenia e incremento en el riesgo de infección y úlceras en la boca o los intestinos por el mal funcionamiento de los neutrófilos (en el tipo Ib).
En la Pubertad:
- Retraso de la pubertad y desarrollo insuficiente.
- Nivel elevado de ácido úrico que puede provocar episodios de gota [14].
- Adenomas hepáticos, que si no se tratan adecuadamente pueden derivar en malignos [15].
- Cálculos renales o insuficiencia renal [16-17].
- Osteoporosis, como consecuencia de un equilibrio cálcico negativo.
- Proteinuria y microalbuminuria.
Diagnóstico de la Enfermedad
Ante la sospecha de la presencia de GSD-I, debe ponerse en marcha un proceso de diagnóstico que incluirá siempre análisis sanguíneos, así como radiografías de hígado y riñones y pruebas de ultrasonido del hígado con el objeto de detectar posibles anomalías en dichos órganos [18].
En lo referente a los análisis sanguíneos debe resaltarse que la hipoglucemia en ayunas con hiperlipidemia, acidosis láctica y una respuesta disminuida o nula de la glucemia a la adrenalina y al glucagón, sugieren fuertemente el diagnóstico, particularmente si se está ante la presencia de hepatomegalia. Por otra parte, la perfusión intravenosa de galactosa aumenta más el nivel de lactato en sangre que el de glucosa.
En aquellos casos en los que no se cuente con un estudio genético previo, el diagnóstico definitivo de la enfermedad de Von Gierke se lleva a cabo mediante la determinación de los niveles de la enzima G-6-Fosfatasa y la presencia de depósitos de glucógeno en el hígado a partir del análisis bioquímico y microscópico de una biopsia hepática. Se deberá proceder de forma urgente con la extracción de la biopsia si los síntomas, los análisis de laboratorio y el examen de los órganos afectados sugieren la presencia de la glucogenosis tipo I. Si existen antecedentes en la familia que hayan desembocado en la realización de estudios genéticos tendentes a identificar las mutaciones de los padres, entonces es posible diagnosticar la enfermedad en nuevos afectados de una forma rápida, precisa y no invasiva, mediante un análisis de ADN, a partir de una muestra sanguínea del paciente, que confirmará la enfermedad si se advierte la presencia simultánea de las mutaciones previamente detectadas en los padres.

Diagnóstico Prenatal y Análisis Genético
Las técnicas utilizadas para el diagnóstico prenatal de otros trastornos, como el estudio enzimático de las vellosidades coriónicas o del líquido amniótico, no sirven para detectar el déficit de la enzima Glucosa-6-fosfatasa, por lo que el diagnóstico prenatal vía análisis enzimático se complica enormemente para la glucogenosis tipo I. En principio, para el subtipo Ia, sí es posible realizar un diagnóstico prenatal a partir de una biopsia del hígado fetal. En la práctica, sin embargo, es una opción poco factible, debido a las dificultades inherentes a la extracción de una biopsia hepática suficientemente grande en un feto, a que la misma tendría que tener lugar en un estado avanzado del embarazo, y a que, además, para la GSD-Ia está demostrada una débil presencia de la Glucosa-6-fosfatasa en el hígado y riñones fetales, lo que podría dar lugar a equívocos en el diagnóstico bioquímico [19-20].
Sin embargo, el diagnóstico prenatal puede resultar factible, para los diferentes subtipos de la GSD-I, mediante un análisis genético del líquido amniótico o de las vellosidades coriónicas, siempre que existan antecedentes familiares que hayan permitido detectar las mutaciones causantes de la enfermedad [21]. Hasta mediados de la década de los noventa no fue posible la identificación del gen responsable de la síntesis de la G-6-fosfatasa, el cual se encuentra localizado en el cromosoma diecisiete (17q21), para el tipo Ia [22]. En la actualidad se han descrito ya amplios listados de mutaciones genéticas que originan la enfermedad de Von Gierke en sus subtipos Ia y Ib [23-27], lo cual denota cierta heterogeneidad genética en esta patología. Estos avances abren, por tanto, la puerta a múltiples aplicaciones, tanto en el ámbito prenatal como postnatal. De esta manera, surge también la posibilidad de un diagnóstico genético preimplantacional como alternativa que ahora es técnicamente posible, a pesar de lo cual todavía no se ha llevado a cabo en nuestro país.
Tratamiento
Hasta la fecha, el tratamiento de la enfermedad de Von Gierke se lleva exclusivamente a cabo mediante terapias paliativas que, principalmente a través del seguimiento de unas pautas nutricionales adecuadas, suelen permitir un control aceptable de la sintomatología de la enfermedad [28-39]. No deben desdeñarse, sin embargo, los avances que se produzcan en el campo de otras incipientes terapias de investigación, tales como la substitución enzimática y las terapias génicas, que podrían proporcionar una cura definitiva de la enfermedad en el transcurso de los próximos años.
Terapias Paliativas
Puede distinguirse entre las terapias paliativas comunes a los tipos Ia y Ib y entre aquellas exclusivas del tipo Ib. Las primeras se refieren principalmente a aspectos nutricionales, mientras que las segundas se centran en el control y la prevención de episodios infecciosos.
Terapias Comunes a los Tipos Ia y Ib
Las metas primordiales del tratamiento de la enfermedad de Von Gierke mediante pautas nutricionales son la prevención de episodios de hipoglucemia que pudieran poner en peligro la vida del paciente, así como la conservación del hígado en las mejores condiciones posibles con el objeto de evitar un posible transplante hepático, ya que este sería una opción a evitar, en la medida de lo posible, y, en cualquier caso, el último de los recursos al que acudir en el tratamiento de esta patología.

El objetivo principal de las terapias nutricionales consiste en reducir al mínimo la acidosis orgánica y conservar los niveles de glucemia por encima de los 70 mg/dL, evitando con ello la hipoglucemia secundaria a una ingesta insuficiente de glucosa.
En el caso de producirse convulsiones hipoglucémicas, será necesario no solo recuperar los niveles normales de azúcar en sangre, sino también, recuperar el equilibrio metabólico mediante la administración de aminoácidos por vía intravenosa.
Se recomienda, por tanto, la administración de alimentos de elevado contenido en almidón separados por intervalos de 2 a 3 horas durante el día, según la tolerancia individual de cada paciente, que puede combinarse con la ingestión de almidón de maíz, y complementarse con la administración nocturna por sonda de preparados líquidos que contengan polímeros de glucosa. Está demostrado que este tratamiento logra mejorar la supervivencia y corregir los trastornos del crecimiento y el desarrollo. Tras el inicio del tratamiento, puede verse un brote de crecimiento de hasta un cm/mes durante el primer año.
La respuesta del paciente a este tratamiento alimenticio es variable, y aunque suelen mejorar significativamente la mayoría de los trastornos asociados a la GSD I, estos no secorrigen por completo. Los niveles de lactato, ácido úrico y triglicéridos tienden a permanecer entre leve y moderadamente elevados en la mayoría de los pacientes.
Alimentación por vía oral. El paciente debe ingerir pequeñas comidas de elevado contenido en almidón cada 2 a 3 horas, o con la frecuencia necesaria para mantener su nivel de glucemia por encima de 70 mg/dL. La primera comida del día debe producirse treinta minutos antes o inmediatamente después de que el paciente interrumpa la alimentación nocturna por sonda, debido al rápido desencadenamiento de hipoglucemia que puede ocurrir tras la detención de la alimentación intragástrica.

Se administran entre cinco y seis comidas al día por vía oral, dependiendo de la duración de la pauta de alimentación por sonda y de las necesidades individuales de cada niño. La última comida debe producirse durante el periodo de dos o tres horas previo al inicio de la alimentación nocturna. La alimentación por vía oral aporta un 60-70% de las kilocalorías en forma de hidratos de carbono, 25-35% en forma de grasa y 10-15% en forma de proteínas. La fuente de hidratos de carbono debe ser fundamentalmente el almidón. Es preciso limitar o evitar el consumo de alimentos que contengan sacarosa, galactosa y fructosa (como las frutas, el azúcar de mesa y la leche y derivados) puesto que estos azúcares se convierten rápidamente en lactato y contribuyen poco o nada a una ingesta constante y adecuada de glucosa.
Debe subrayarse la importancia de una pauta frecuente de alimentación y de la ingestión de alimentos con elevado contenido en almidón. La incorporación de cierta cantidad de proteínas y grasas a cada comida contribuye a prolongar el periodo de absorción de la glucosa.
Alimentación nocturna por sonda. La alimentación nocturna por sonda precisa de la administración de glucosa exógena a una velocidad que reduzca la necesidad hepática de producir glucosa, obviando así de forma eficaz la función fundamental de la G-6-Fosfatasa ausente. La mayoría de los lactantes y niños normales produce glucosa a una velocidad de 5 a 8 mg/kg de peso corporal/minuto. Para los afectados por la enfermedad de Von Gierke el ajuste de la velocidad de alimentación por sonda -para alcanzar un nivel igual o ligeramente superior a la velocidad normal de producción hepática de glucosa – resulta un factor fundamental en el control de los niveles sanguíneos de lactato.
La administración de glucosa a una velocidad de 8 a 9 mg/kg de peso corporal/minuto previene la hipoglucemia y reduce al mínimo la acidosis orgánica en la mayoría de los pacientes con GSD-I. Es preciso modificar la velocidad de administración de forma individualizada, revaluándola cada tres a seis meses. Toda velocidad de infusión superior a la indicada puede producir anorexia diurna, incapacidad de consumir cantidades adecuadas de hidratos de carbono durante el día e ingesta insuficiente de proteínas.
Resulta esencial aportar el preparado mediante infusión regular y constante, empleando una bomba de infusión con sistema de alarma que indique oclusión de la sonda o fallos de la bomba. La hipoglucemia que sigue a la interrupción de la alimentación por sonda es mucho más rápida que la que se produce después de una comida. En algunos casos de detención accidental de la infusión los pacientes han fallecido a consecuencia de una hipoglucemia rápida y grave.
En el caso de que el niño se quite la sonda durante la noche, no serían efectivas las alarmas de la bomba de infusión, ya que no hay oclusión, y la mejor forma de evitar esto es utilizando un “empapador con alarma”. Se puede ver un modelo en http://www.nitetrain-r.com.
La mayoría de los niños puede aprender a introducirse su propia sonda nasogástrica por la noche sin dificultad. Es preciso instruir a padres e hijos cuidadosamente sobre el uso y cuidado de la bomba. Si el uso de la sonda nasogástrica no es viable, debe considerarse la implantación de una sonda de gastrostomía. Sin embargo, con la preparación adecuada, es posible tratar a la mayoría de los pacientes mediante sonda nasogástrica u orogástrica. En los niños con GSD-Ib, debido a su neutropenia, la gastrostomía es muchas veces causa de problemas por las infecciones del estoma, por lo que, a diferencia de otras enfermedades, en este caso sería recomendable intentar la alternativa de la sonda nasogástrica siempre que sea posible.
Tratamiento con almidón de maíz. En niños mayores, adolescentes y adultos, puede emplearse el almidón de maíz no cocido (MaizenaR) como alternativa de tratamiento eficaz para pacientes con GSD-I. Se ha demostrado que la ingestión de 1,75-2,5 gr. de almidón de maíz por kilogramo de peso corporal cada seis horas, que aporta 5,3-7,6 mg de glucosa por kg de peso corporal y minuto, mantiene una glucemia relativamente constante, siempre que la glucemia inicial fuese normal. El tratamiento a largo plazo con almidón de maíz ha resultado tan eficaz como la alimentación nocturna mediante sonda nasogástrica en cuanto a conservación constante de los niveles de glucemia y restitución del crecimiento normal.
Inicialmente no se recomendaba el tratamiento con almidón de maíz para los lactantes o los niños pequeños, ya que se creía que niveles propios del adulto de amilasa pancreática – una de las dos enzimas necesarias para la hidrólisis del almidón -, no se alcanzan hasta los dos-cuatro años. Sin embargo, en la actualidad la maizena se introduce antes de la edad de dos años, e incluso se ha utilizado con éxito en niños de 8 meses.
La preparación adecuada del almidón de maíz resulta indispensable para un tratamiento adecuado. Es preciso preparar el almidón de maíz con agua a temperatura ambiente. Los efectos colaterales típicos (diarrea transitoria, distensión abdominal y meteorismo) son poco importantes y se resuelven de forma espontánea.
Es importante introducir de forma paulatina el tratamiento con almidón de maíz, utilizando concentraciones crecientes a medida que avanza el tratamiento, con el objeto de “madurar” el sistema enzimático del páncreas; aún así hay niños que no responden a la terapia del almidón de maíz.
Últimamente se está desarrollando un nuevo tipo de almidón modificado, cuyos primeros resultados en adolescentes afectados con glucogenosis tipo Ia y Ib son bastante alentadores, aumentando aún más el periodo de ayuno nocturno, en comparación con el almidón de maíz ya conocido, y presentando menos complicaciones intestinales tras su ingesta vía oral o por sonda gástrica. De todos modos, aún no está comercialmente disponible debido a que aún se encuentra en fase de pruebas.
Tratamiento con Glycosade. Inicialmente, en el añoo 2002 varios grupos de investigación (respectivamente liderados por los Dres. Karen Kumor, Ralph Waniska, David Weinstein, Phil Lee y Kaustuv Batycharria, estos dos últimos de Londres, Reino Unido) comenzaron a buscar un nuevo sustituto del almidón de maíz crudo, que permitiese espaciar más los tiempos de ingestión, especialmente durante el horario nocturno, con el propósito de alargar los periodos de descanso de los afectados por glucogenosis tipo I. Finalmente el grupo de los Dres. Lee y Batycharria identificaron el mejor candidato, denominado Glycosade y desarrollado por Vitaflo International Ltd. Entre el 2006 y 2007, el equipo del Dr. Weinstein realizó los primeros estudios sobre la eficacia del producto en 12 voluntarios, con glucogenosis tipos Ia y Ib, durante el periodo nocturno y se encontró una mejora estadísticamente significativa en el mantenimiento de la normoglucemia durante más tiempo, en comparación con el tradicional almidón de maíz [40]. Este nuevo sustituto del almidón de maíz, Glycosade, fue aprobado para su uso en Inglaterra, Australia, Francia y Alemania. En EE.UU. y en España todavía esta pendiente de aprobación.
Entre el 2008 y el 2009, más de 30 pacientes con tipo Ia comenzaron a participar en nuevos estudios realizados por las universidades de Duke y Florida (EE.UU.) para optimizar la dosis de uso más adecuada para estos pacientes.
Los Triglicéridos de Cadena media (TCM). Desde hace años se propuso utilizar suplementos de estos ácidos grasos con la idea de mejorar el control metabólico y el desarrollo de los pacientes, aunque los resultados no fueron concluyentes. En un artículo, publicado por el Dr. Das y colaboradores en el año 2010 [55], se relata una buena respuesta en 2 pacientes tratados con suplementos de TCM en cuanto a los niveles de Ácido Úrico, Triglicéridos y crecimiento.
El transplante hepático. El transplante, hoy en día, solo debe considerarse en aquellos pacientes que no respondan a un tratamiento dietético adecuado o que hayan desarrollado adenomas malignos. Corrige la enfermedad tanto en el tipo Ia como en el Ib; sin embargo, en este último no corrige la neutropenia [41-43].
Debe resaltarse, en cualquier caso, que, en el transcurso de las dos últimas décadas, se ha podido contrastar de manera fehaciente que, en el tratamiento de pacientes con glucogenosis tipo I, la infusión nasogástrica nocturna (o las tomas de maicena cruda) asociada a una ingesta de comidas frecuentes durante el día, contribuye a la prevención o regresión de los adenomas hepáticos.
Tratamiento y prevención de la enfermedad renal. Últimamente se está dando una mayor importancia a las complicaciones a largo plazo de las Glucogenosis tipo I y entre ellas a la enfermedad renal, de la que se publican trabajos sobre su patogenia basada en el estrés oxidativo [59]. La primera manifestación de la enfermedad renal suele ser la microalbuminuria y su presencia obliga a la administración de Inhibidores de la Enzima Conversora de Angiotensina (IECA) como por ejemplo Enalapril, comenzando con dosis bajas de 2,5 mgr/día.
Terapias Exclusivas del Tipo Ib
Neutropenia. Uno de los aspectos diferenciales en el tipo Ib está en el tratamiento y prevención de las infecciones recidivantes. Las alteraciones de los neutrófilos son independientes del control metabólico, por eso es necesaria la adopción de otro tipo de medidas. Recientes estudios ponen en evidencia la relación entre la neutropenia y la disfunción de los neutrófilos y determinadas mutaciones genéticas y las vías específicas por las que el déficit enzimático afecta a estas células sanguíneas [56,58].
En esta situación, la utilización de GM-CSF (factor recombinante humano estimulante de la colonia de granulocitos macrófagos) y G-CSF (factor estimulante de la colonia de granulocitos) proporciona, en general, buenos resultados. La dosis recomendada de GM-CSF es de 7 mg/kg de peso/día, y la de G-CSF de 3 mg/kg de peso/día, ambas por vía subcutánea [44]. También pueden utilizarse antibióticos de manera profiláctica, habitualmente SeptrimR, aunque, en cualquier caso, el uso preventivo de los mismos puede ser cuestionable y está sujeto a debate.
Enfermedad inflamatoria intestinal (EII). El otro aspecto a destacar es la predisposición a la enfermedad inflamatoria intestinal debido a la neutropenia congénita. Los afectados por esta patología suelen sufrir de diarreas intermitentes, que suelen empeorar con la edad. La causa de esta diarrea todavía se desconoce, aunque se han postulado varias hipótesis: absorción de la glucosa intestinal alterada que daría lugar a diarreas osmóticas, adsorción del almidón de maíz que provocaría inflamación, y absorción intestinal alterada en general como el resultado del depósito de glucógeno en el intestino. El modelo de la EII en la Glucogenosis Ib es muy similar al de los pacientes con enfermedad de Crohn y en un artículo del Dr. Weinstein [57] pone de manifiesto la coincidencia en los hallazgos serológicos entre las dos enfermedades, presentando ambas un elevado porcentaje de positividad a anticuerpos antiflagelina bacteriana, por ello su tratamiento también es común. La incidencia de la enfermedad inflamatoria intestinal en personas afectadas con glucogenosis tipo Ib es alta, como indicó un estudio retrospectivo en donde se encontró una incidencia del 75% [45]. El tratamiento indicado para reducir estos síntomas se basa en la administración de anti-inflamatorios intes-tinales, así como principios activos como la Mesalazina (PentasaÒ), la Sulfasalazina (SalazopyrinaÒ) u otros equivalentes se suelen administrar a las dosis habituales.
Terapias en Fase de Investigación
En el campo de la enfermedad de Von Gierke, tanto la terapia de substitución enzimática como la terapia génica se encuentran en estadios de investigación aún muy incipientes. Cabe esperar, sin embargo, que a medio y largo plazo, estas terapias den lugar a tratamientos más efectivos para la GSD-I, de igual forma que las mismas ya han alcanzado cierto desarrollo en el ámbito de otras enfermedades raras, o incluso para otros tipos de glucogenosis, como la glucogenosis tipo II o enfermedad de Pompe.
Terapia enzimática. Consiste en reponer en el hígado la enzima que falta (G-6-Fosfatasa). El problema que se presenta, en este caso, es que resulta muy difícil hacer llegar una enzima producida en laboratorio al lugar adecuado de la célula, para que allí realice su función. Esto es así porque el complejo enzimático de la Glucosa-6-Fosfatasa no está en el torrente circulatorio, ni libre en el citoplasma, sino que forma parte de la membrana del retículo endoplasmático, concretamente de la cara interna de dicha membrana. Esta dificultad añadida complica enormemente la viabilidad terapéutica de la substitución enzimática en el tratamiento de la glucogenosis tipo I, ya que no bastaría con administrar la enzima a los afectados, sino que, además, habría que conseguir “instalarla” en su lugar adecuado. Hasta la fecha, no se han logrado resultados aceptables en este campo, situación que se ve enormemente dificultada por los escasos recursos económicos que se dedican a promover la investigación en una enfermedad catalogada como rara, y por tanto, considerada poco rentable.
Terapia génica. Consiste en preparar el gen en laboratorio y luego colocarlo en el lugar adecuado para que sintetice la enzima G-6-fosfatasa. La dificultad, en este caso, estriba en conseguir que los genes utilizados se mantengan en un nivel terapéutico y por un tiempo prolongado y, por otra parte, en prevenir la respuesta del sistema inmunológico para que esta no impida la transferencia de los genes.
De cualquier modo, se han obtenido resultados prometedores en ratones glucogénicos, a los que se les ha infundido un vector adeno-vírico portador del gen de la Glucosa 6-fosfatasa. De esta forma, se ha conseguido en los ratones tratados un incremento en su actividad enzimática, con disminución de los depósitos de glucógenos en hígado y riñón, descenso de los niveles sanguíneos de triglicéridos, ácido úrico y colesterol, y elevación de la glucosa [46-50].
Últimamente también a perros genéticamente predispuestos a sufrir la enfermedad se les ha infundido un vector adenovírico (AAV) portador del gen de la Glucosa -6-fosfatasa. En un primer estudio, se administró una primera variante del vector adenovírico, AAV2/8, a tres perros con la glucogenosis tipo Ia de edad superior a 11meses. Los valores de los marcadores en orina, incluyendo el lactato y el 3-hidroxibutirato, se corrigieron inducidos por la expresión del gen de la glucosa-6-fosfatasa hepática. La acumulación de glucógeno en el hígado se redujo casi a valores normales en estos perros tratados [51]. En otro trabajo más reciente, se administró una nueva generación de adenovirus asociado (rAAV2/8), mejorada con respecto a la anteriormente mencionada, se le administró a otro perro afectado con la tipo Ia en su primer día de vida y a las dos semanas se observó una clara mejoría en los síntomas de la enfermedad. La mejora fue transitoria puesto que, dos meses después del tratamiento, el perro tratado con dicho adenovirus mejorado ya no podía mantener los niveles normales de glucosa en sangre después de una hora de ayuno.
El mismo animal se dosificó a continuación, con otro vector terapéutico (rAAV2/1) infundido a través de la vena porta. Dos meses después de administrar la dosis del vector rAAV2/1, tanto los niveles de glucosa en sangre como de lactato fueron normales tras 4 horas de ayuno. Con ayunos más prolongados, el perro sigue manteniendo las concentraciones de glucosa cercanos a la normalidad, aunque los niveles de lactato se mantuvieron elevados durante 9 horas. La suplementación de glucosa a través de la dieta se suspendió a partir del mes siguiente al de la administración del vector adenovírico rAAV2/1 y el perro continúa creciendo con mínimas alteraciones en los valores clínicos de referencia tras 23 meses (18 meses después del tratamiento con rAAV2/1) [52].
Esta terapia se presenta como una de las grandes esperanzas de cara a la futura curación de esta enfermedad, y buena parte de los trabajos de investigación recientes sobre la enfermedad de Von Gierke se han centrado en dicho campo.
Terapia celular. Consiste en introducir en el hígado del individuo, afectado con la glucogenosis tipo I, células madre hepáticas de un individuo sano adulto, lo que conlleva menos riesgo que un transplante hepático, y los primeros resultados observables demuestran una corrección en los niveles de glucosa sanguínea, con ausencias de hipoglucemia e incluso posibilidad de tener una dieta y vida normales [53]. Recientemente, en el Hospital La Fe de Valencia se ha iniciado un programa de Trasplante de Hepatocitos como una nueva opción terapéutica para el manejo de niños con determinadas enfermedades metabólicas como las glucogenosis. La técnica que emplean consiste en infundir hepatocitos aislados a partir de hígados no aptos para transplante de órgano completo por la vena porta para que se implanten en el hígado receptor. En el Hospital La Fe se ha tratado una niña con glucogenosis Ia.La paciente es una niña de 6 años con un mal control metabólico. Tras reajuste dietético y el trasplante de hepatocitos la niña no ha presentado ninguna hipoglucemia sintomática, ha mejorado su acidosis, mantiene una adecuada curva pondo estatural y ha iniciado escolarización mejorando de manera importante su calidad de vida. Esta niña no ha tenido ninguna complicación durante el procedimiento.
Aunque se necesitan hacer más ensayos para comprobar la eficacia de esta terapia, no cabe duda que es una nueva posibilidad en el camino de búsqueda de una terapia curativa de la enfermedad.
Para más información: aquellos profesionales de la salud interesados en obtener más información sobre la enfermedad y su tratamiento, pueden ponerse en contacto con el siguiente profesional, debido a su acreditada experiencia con pacientes afectados por la glucogenosis tipo I:
Dr. Leopoldo Garcia Alonso
Jefe de la Unidad de Gastro-Nutricion Pediatrica
Complejo Hospitalario Universitario Juan Canalejo
As Xubias, 84
15006 La Coruna
Teléfono: 981 17 80 00
Referencias Bibliográficas
[1] Hers H et al (1989) “Glycogen storage diseases”, en Scriver CR et al. eds. The metabolic basis of inherited disease. 6th ed. New York. McGraw-Hill; pp. 425-452.
[2] Reis CV et al (1999) “Glicogenose tipo I”, Jornal de Pediatria; 75 (4): 227-236
[3] Von Gierke EO (1929) “Glykogenspeicherkrankheiter leber und Nieren”, Beitrage zur Pathologischen Anatomie und zur Allgemeinen Pathologie; 82: 497-513.[4] McKusick, VA ed. (2004) Online mendelian inheritance in man (OMIM). Baltimore. The Johns Hopkins University. Entry no 232200.
[5] Senior B y L Loridan (1968) “Functional differentiation of glycogenoses of the liver with respect to the use of glycerol”, New England. Journal of Medicine; 279: 965-970.
[6] Chen YT et al (1988) “Renal disease in type I glycogen storage disease”, New England Journal of Medicine; 318: 7-11.
[7] Chen YT y JL Van Hove (1995) “Renal involvement in type I glycogen storage disease”, Advances in Nephrology from the Necker Hospital; 24: 357-365.
[8] Kim SY et al (2007). “Neutrophilia and elevated serum cytokines are implicated in glycogen storage disease type Ia”. FEBS Letters, 581: 3833-3838.
[9] Kim SY et al (2008). “Hepatic injury correlates with increased neutrophil infiltration of the liver in glycogen storage disease Type Ia”. Journal of Hepatology, 48: 479-485.
[10] Guven AG et al (2006) “Severe lactic acidosis and nephrolithiasis in an infant etiology?: type 1 glycogen storage disease (GSD)”, Pediatric Nephrology; 21 (6): 761-765.
[11] Badolato R et al (2004) “Congenital neutropenia: advances in diagnosis and treatment”, Current Opinion in Allergy & Clinical Immunology; 4 (6): 513-521.
[12] Smit GP et al (1993) “The long-term outcome of patients with glycogen storage disease type Ia”, European Journal of Pediatrics; 152: S52-S55.
[13] Moraru E et al (2007) “Glycogen storage disease type I bet-ween chronic ambulatory follow-up and pediatric emergency”, Journal of Gastrointestinal and Liver Diseases; 16 (1): 47-51.
[14] Hou JW et al (1996) “Glycogen storage disease type Ia (Von Gierke Disease) complicated by gouty arthritis and xanthomatosis”, Archives of Pediatric and Adolescent Medicine; 150: 219-20.
[15] Franco LM et al (2005) “Hepatocellular carcinoma in glycogen storage disease type Ia: a case series”, Journal of Inherited Metabolic Disease; 28 (2): 153-162.108
[16] Lin CC et al (2005) “Renal sonographic findings of type I glycogen storage disease in infancy and early childhood”, Pediatric Radiology; 35 (8):786-791.
[17] Hara T et al (2007) “Unsuccessful management for renal failure induced by glycogen storage disease type-I (Von Gierke disease) in peritoneal dialysis”, Nippon Naika Gakkai Zasshi; 96 (4): 775-777.
[18] Burchell A y ID Waddell (1990) “Diagnosis of a novel glycogen storage disease: type IaSP”, Journal of Inherited Metabolic Diseases;13: 247-249.
[19] Golbus M et al (1988) “The prenatal determination of glucose-6-phosphatase activity by fetal liver biopsy”, Prenatal Diagnosis; 8: 401-404.
[20] Pan CJ et al (1998) “Ontogeny of the murine glucose-6-phosphatase system”, Archives of Biochemistry and Biophysics; 358: 7-24.
[21] Lam CW et al (2000) « Prenatal diagnosis of glycogen storage disease type Ib using denaturing high performance liquid chromatography”, Prenatal Diagnosis; 20:765-768.
[22] Lei KJ et al (1993) “Mutations in the glucose-6-phosphatase gene that cause glycogen storage disease type 1a”, Science; 262 (5133): 580-583.[23] Chou JY et al (2002) “Type I Glycogen Storage Diseases: Disorders of the Glucose-6-Phosphatase Complex”, Current Molecular Medicine; 2: 121-143.
[24] Hidaka F et al (2005) “A novel mutation of the PHKA2 gene in a patient with Xlinked liver glycogenosis type 1”, Pediatrics International; 47 (6): 687-90.
[25] Han SH et al (2005) “A novel mutation (A148V) in the glucose 6-phosphate translocase (SLC37A4) gene in a Korean patient with glycogen storage disease type 1b”, Journal of Korean Medical Science; 20 (3): 499-501.
[26] Melis D et al (2005) “Genotype/phenotype correlation in glycogen storage disease type 1b: a multicentre study and review of the literature”, European Journal of Pediatrics; 164 (8): 501-508.
[27] Lam CW et al (2006) “Resequencing the G6PT1 gene reveals a novel splicing mutation in a patient with glycogen storage disease type 1b”, Clinica Chimica Acta; 374 (1-2): 147-148.
[28] Greene HL et al (1976) “Continuous nocturnal intragastric feeding for management of type 1 glycogen-storage disease”, New England Journal of Medicine; 294: 423-425.
[29] Folk CC y HL Greene (1984) “Dietary management of type I glycogen storage disease”, Journal of the American Dietary Association; 84: 293-301.
[30] Chen YT et al (1984) “Cornstarch therapy in type I glycogen-storage disease”, New England Journal of Medicine; 310: 171-175.
[31] Stanley CA (1985) “lntragastric feeding in glycogen storage disease and other disorders of fasting”, en: Walker WA y JB Watkins JB eds. Nutrition in Pediatrics. Boston. Little-Brown; pp: 781-791.
[32] Haymond MW y WF Schwenk (1986) “Optimal rate of enteral glucose administration in children with glycogen storage disease type I”, New England Journal of Medicine; 314: 682-685.
[33] Folk C y S Rarback (1988) “Dietary management of glycogen storage disease type I”, Top Clinical Nutrition ; 3 (4):77-81.
[34] Smit GP et al (1988) “Complex carbohydrates in the dietary management of patients with glycogenosis caused by glucose-6-phosphatase deficiency”, American Journal of Clinical Nutrition; 48 (1): 95-97.
[35] Wolfsdorf JI et al (1990) “Continuous glucose for treatment of patients with type I glycogen-storage disease: comparison of the effects of dextrose and uncooked cornstarch on biochemical values”, American Journal of Clinical Nutrition; 52:1043-1050.
[36] Wolfsdorf JI et al (1990) “Glucose therapy for glycogenosis type I in infants: comparison of intermittent uncooked cornstarch and continuous overnight glucose feedings”, Journal of Pediatrics; 117: 384-391.
[37] Hayde M y K Widhalm (1990) “Effects of cornstarch treatment in very Young children with type I glycogen storage disease”, European Journal of Pediatrics; 149:630-633.
[38] Johnson MP et al (1990) “Metabolic control of Von Gierke disease (glycogen storage disease type IA) in pregnancy: maintenance of euglycemia with cornstarch”, Obstetrics and Gynecology; 75: 507-510.
[39] Wolfsdorf JI y F Crigler (1997) “Cornstarch regimens for nocturnal treatment of young adults with type I GSD”, American Journal of Clinical Nutrition; 65:1507-1511.
[40] Correia CE, et al. (2008) “Use of modified cornstarch thera-py to extend fasting in glycogen storage disease types Ia and Ib”, American Journal of Clinical Nutrition; 88 (5): 1272- 1276.
[41] Muraca M y AB Burlina (2005) “Liver and liver cell transplantation for glycogen storage disease type IA”, Acta Gastroenterologica Belgica; 68 (4): 469-472.
[42] Martin AP et al (2006) “Successful staged kidney and liver transplantation for glycogen storage disease type Ib: A case report”, Transplantation Proceedings; 38 (10): 3615-3619.
[43] Carreiro G et al (2007) “Orthotopic liver transplantation in glucose-6-phosphatase deficiency–Von Gierke disease–with multiple hepatic adenomas and 110 concomitant focal nodular hyperplasia”, Journal of Pediatric Endocrinology and Metabolism; 20 (4): 545-549.
[44] De Diego Fernandez P et al (2001) “Tratamiento continuo con factores estimulantes de colonias (G-CSF) de la neutropenia asociada a la glucogenosis tipo Ib”, Anales Españoles de Pediatría; 55: 282-284.
[45] Visser G et al (2000) “Neutropenia, neutrophil dysfunction, and inflammatory bowel disease in glycogen storage disease type Ib: results of the European Study on Glycogen Storage Disease type I”, Journal of Pediatrics; 137 (2):187-91.
[46] Chou JY et al (2002) “Adenovirus-mediated gene therapy in a mouse model of glycogen storage disease type 1a”, European Journal of Pediatrics; 161: S56-S61.
[47] Koeberl DD et al (2006) “Early, sustained efficacy of adeno-associated virus vector-mediated gene therapy in glycogen storage disease type Ia”, Gene Therapy; 13 (17): 1281-1289. Erratum en: Gene Therapy; 13 (19): 1430 y Gene Therapy; 14 (3):281.
[48] Ghosh A et al (2006) “Long-term correction of murine glycogen storage disease type Ia by recombinant adeno-associated virus-1-mediated gene transfer”, GeneTherapy; 13 (4): 321-329.
[49] Chou JY y BC Mansfield (2007) “Gene therapy for type I glycogen storage diseases”, Current Gene Therapy; 7 (2): 79-88.
[50] Koeberl DD et al (2007) “Efficacy of helper-dependent adenovirus vectormediated gene therapy in murine glycogen storage disease type Ia”, Molecular Therapy; 15 (7):1253-1258.
[51] Koeberl DD et al. (2008) “AAV vector-mediated reversal of hypoglycemia in canine and murine glycogen storage disease type Ia”, Molecular Therapy; 16 (4): 665-672.
[52] Weinstein DA et al. (2010) “AAV-mediated correction of a canine model of glycogen storage disease type Ia”, Hum Gene Ther; (pendiente de impresion).
[53] Lee KW et al. (2007) “Hepatocyte transplantation for glycogen storage disease type Ib”, Cell Transplantation; 16(6):629-637.
[54] Cassiman D et al. “An adult male patient with multiple adenomas and a hepatocellular carcinoma: mild glycogen storage disease type Ia”, J Hepatol. 2010 Jul;53(1):213-7.
[55] Das AM. et al. ”Glycogen storage disease type 1: impact of medium-chain triglycerides on metabolic control and growth”.Ann Nutr Metab. 2010;56(3):225-32.
[56] Hayee B et al.” G6PC3 mutations are associated with a major defect of glycosylation: a novel mechanism for neutrophil dysfunction” Glycobiology. 2011 Mar 8.111
[57] Davis MK.et al.“Antibodies to CBir1 are associated with glycogen storage disease type Ib.”J Pediatr Gastroenterol Nutr. 2010 Jul;51(1):14-8.
[58] Jun HS, et al.”Lack of glucose recycling between endoplasmic reticulum and cytoplasm underlies cellular dysfunction in glucose-6-phosphatase-beta-deficient neutrophils in a congenital neutropenia syndrome.” Blood. 2010 Oct 14;116(15):2783-92.
[59] Yiu WH et al.”Oxidative stress mediates nephropathy in type Ia glycogen storage disease. Lab Invest. 2010 Apr;90(4):620-9.

Más notas de la edición 16
Lee desde Issuu nuestra última edición publicada en Octubre 2025, Edición número 170

Notas relacionadas a Glucogenosis Tipo I: Enfermedad de Von...

