



Descripción de las principales alteraciones epigenéticas asociadas con el desarrollo de cáncer colorrectal, próstata, mama y gástrico: Revisión de tema
Andrés-David López-Estupiñán1,a, Andrés-Felipe Mondragón-Cháves1,a, Andrés-Felipe Giraldo-Velásquez1,a, Juan-David Olave-Medina1,a, Elizabeth Londoño-Velasco2,a
1 Estudiante de medicina. Semillero de Innovadores en Salud ISSEM.
2 Bióloga, Magíster en Ciencias Biomédicas, Profesora Departamento de Ciencias Básicas de la Salud.
a Facultad de Ciencias de la Salud, Pontificia Universidad Javeriana Cali (Colombia)
Correspondencia: Andrés David López Estupiñán – ORCID ID https://orcid.org/0000-0001-7796-6370 – Facultad de Ciencias de la Salud – Pontificia Universidad Javeriana Cali (Colombia) – E-mail:andresle98@outlook.com
Conflicto de intereses: Los autores del artículo hacen constar que no existe, de manera directa o indirecta, ningún tipo de conflicto de intereses que pueda poner en peligro la validez de lo comunicado. López-Estupiñán AD, Mondragón-Chávez AF, Giraldo-Velásquez AF, Olave-Medina JD, Londoño-Velasco E. Descripción de las principales alteraciones epigenéticas asociadas con el desarrollo de cáncer colorrectal, próstata, mama y gástrico: Revisión de tema. Salutem Scientia Spiritus 2021; 7(1):36-51.
Resumen
Actualmente, los marcadores epigenéticos son reconocidos en el área clínica por su potencial utilidad en momentos de diagnóstico, pronóstico y predicción a la respuesta al tratamiento con antineoplásicos en diferentes tipos de cáncer. Por tal razón, en esta revisión de tema se describen las principales vías moleculares y cambios epigenéticos asociados con el desarrollo y progresión de los cuatro tipos de cáncer de mayor incidencia en el departamento del Valle del Cauca (colorrectal, próstata, mama y gástrico), con el propósito de vislumbrar la aplicación de pruebas que permitan el análisis de biomarcadores epigenéticos de tumoración en la práctica clínica. Para ello se realizó una búsqueda bibliográfica de artículos científicos publicados entre los años 2004-2019 en las bases de datos de Medline y Cochrane Library. Dependiendo del tipo de cáncer se reconocieron las siguientes alteraciones epigenéticas predominantes. En colorrectal el fenotipo de metilación de islas CpG principalmente por moléculas MLH1 y p16 permite clasificar el tipo de cáncer colorrectal presente, y el RNA no codificante miR-133a es el que define el paso de adenoma avanzado a adenocarcinoma, de igual manera se debe resaltar eventos como la hipometilación genómica global. En el cáncer de próstata principalmente se vio involucrada la hipermetilación y el silenciamiento de la región promotora del gen GSTP1, amplificación genética de histona metiltransferasa EZH2, y los patrones aberrantes de H3 y H4. De igual manera en el cáncer de mama se vio la hipermetilación de las islas CpG promotoras de los genes BRCA1 y RASSF1, la sobreexpresión de receptores tipo estrógenos, y se resalta el silenciamiento de moléculas de adhesión tipo E-Cadherina en periodos de crecimiento tumoral y metástasis. En cáncer gástrico la infección por H. pylori genera un estado de hipometilación en regiones encargadas del crecimiento y la proliferación celular, también se vio la hipermetilación de las islas CpG de los genes hMLH1 y p14/ARF. A grandes rasgos un evento predominante encontrado en todos los tipos de cáncer fue la hipermetilación de DNA, llevando al silenciamiento moléculas indispensables en la regulación del crecimiento y proliferación celular.
Palabras clave: Marcadores epigenéticos, neoplasias colorrectales, neoplasias de la mama, neoplasias gástricas, neoplasias de la próstata.
Abstract: Description of the main epigenetic disturbances associated with the development of colorectal, prostate, breast and gastric cancer: Topic review
Nowadays, the epigenetic markers are recognized in the clinical area for their potential utility at times of diagnosis, prognosis, and prediction of the response to antineoplastic treatment in different types of cancer. For this reason, this topic review describes the main molecular pathways and epigenetic changes associated with the development and progression of the four types of cancer with the highest incidence in the department of Valle del Cauca (colorectal, prostate, breast and gastric), with the purpose of envisioning the application of tests that allow the analysis of epigenetic biomarkers of tumors in clinical practice .For this reason, a bibliographic search of scientific articles published between the years 2004 – 2019 was carried out in the databases of Medline and Cochrane Library. Depending on the type of cancer, the following predominant epigenetic alterations were recognized. In colorectal the phenotype of CpG islands methylation mainly by MLH1 and p16 molecules allows classifying the type of colorectal cancer that is present, and the miR-133a non-coding RNA is what defines the advanced adenoma to adenocarcinoma passage. Similarly, events such as global genomic hypomethylation should be highlighted in the progression of this cancer. In prostate cancer, hypermethylation and silencing of the promoter region of the GSTP1 gene, genetic amplification of histone methyltransferase EZH2, and aberrant patterns of H3 and H4 were mainly involved. Similarly, in breast, hypermethylation of the promoter CpG islands of the BRCA1 and RASSF1 genes, overexpression of estrogen-like receptors, and highlighting the silencing of E-Cadherin-like adhesion molecules during periods of tumor growth and metastasis were seen. In gastric cancer, H. pylori infection generates a hypomethylation state in regions responsible for cell growth and proliferation, hypermethylation of the CpG islands of the hMLH1 and p14 / ARF genes was also seen. Broadly speaking, a predominant event found in all types of cancer was DNA hypermethylation, leading to the silencing of molecules that are essential in the regulation of cell growth and proliferation.
Keywords: Epigenomic markers, Colorrectal neoplasms, Breast neoplasms, Stomach neoplasms, Prostatic neoplasms.
Introducción
El cáncer es una de las principales causas de morbilidad y mortalidad en el mundo. Su detección en fase avanzada, la falta de diagnóstico y tratamiento oportuno, identifican a esta patología como una problemática de interés mundial no sólo para la salud pública, sino también para el conocimiento básico de las ciencias biomédicas. Con los años hemos sido testigos de gran número de evidencias experimentales y clínicas que apoyan la idea de la existencia de dos pilares fundamentales para la génesis del proceso cancerígeno. El primero hace referencia a la predisposición genética, la cual es soportada por los patrones de recurrencia y riesgo relativo encontrados en diferentes tipos de cáncer familiar, y el segundo se refiere a la actuación de factores ambientales (cancerígenos físicos, químicos y biológicos) con un alto potencial para alterar la integridad genómica y la función celular. Luego, identificar el origen específico de aquellas alteraciones moleculares y celulares que conllevan a la transformación maligna resulta ser una labor compleja.
Ahora bien, teniendo en cuenta que el cáncer a menudo surge de la acumulación de sucesivos defectos genéticos (mutaciones) y que la tasa de evolución clonal depende de la frecuencia con la cual aparecen nuevas mutaciones que aceleran su progresión. Las mutaciones en genes que regulan el ciclo celular (TP53, RB, MYC), la reparación del DNA (BRCA2, BRCA1, ATM), la telomerasa (TERT, TERC), el metabolismo (GSTP1), entre otros, han sido objeto de un gran número de investigaciones, que reconocen el papel preponderante de estas alteraciones en el desarrollo y progresión de diferentes tipos de cáncer. Según el instituto nacional de cáncer NIH, el 10% de los cánceres pueden ocasionarse por mutaciones genéticas heredadas, a lo cual se asocian más de 50 síndromes hereditarios correspondientes a cáncer. Mientras que la mayoría de los casos de cáncer no se heredan y muchos de ellos están influenciados por factores ambientales, que causan mutaciones somáticas que estimulan la división celular o afectan de alguna otra forma el proceso de progresión de la enfermedad, y es aquí donde la epigenética cobra relevancia.
La epigenética se define como el estudio de los cambios heredables que regulan la expresión de los genes mediante mecanismos distintos a la modificación en la secuencia del DNA, que determinan un fenotipo específico.1 Estos mecanismos desempeñan un papel clave en los procesos fisiológicos, pues repercuten sobre la especificidad en el crecimiento y la diferenciación celular, por tanto, patrones de expresión génica anormales modificarán la identidad celular y aumentarán la susceptibilidad al desarrollo de ciertas enfermedades como el cáncer. La epigenética considera tres mecanismos por los cuales se alteran los patrones de expresión génica:2 la metilación del DNA, las modificaciones químicas sobre las colas de las histonas y la expresión de RNA no codificante. De los anteriores, los cambios en los patrones de metilación del DNA y la modificación de las histonas son los más asociados con cambios en el fenotipo, que culminan con el desarrollo de diferentes tipos de cáncer.3,4 En algunos casos, el DNA de las células cancerosas está hipermetilado, y en otros hipometilado. Su efecto en el desarrollo de cáncer dependerá de la región génica involucrada. Se cree que la hipermetilación contribuye al silenciamiento de genes supresores. Mientras que el efecto de la hipometilación aún no está tan claro.
Existen minuciosas y extensas revisiones que exponen las alteraciones epigenéticas de diferentes tipos de cáncer de manera individual. Sin embargo, son pocas las revisiones que realizan una descripción conjunta de las principales alteraciones epigenéticas encontradas en las neoplasias de mayor incidencia a nivel mundial y regional. Entre los cánceres de mayor incidencia a nivel mundial se encuentran el cáncer de próstata, mama, colorrectal y gástrico. Según la IARC, durante el año 2018 el 11,5% de los nuevos diagnósticos de cáncer correspondió a cáncer de mama (2’088.849), el 10,2% debido a cáncer colorrectal (1’849.518), el 7,1% pertenece a cáncer de próstata (1´276.106) y el 5,7% aportado por cáncer gástrico (1´033.701).5
En Colombia se reportaron 101.893 casos nuevos de cáncer en el 2018, de los cuales un 13,1% corresponde a cáncer de mama (13.380), el 12,5% cáncer de próstata (12.712), 9% cáncer colorrectal (9.140), y 7,3% por cáncer gástrico (7.419).6 Según el Registro Poblacional de Cáncer de Cali, se espera que para el año 2019 aparezcan 3.250 nuevos diagnósticos de cáncer en mujeres, siendo el de mama la mayor morbilidad con 23,3% de los casos (754). En relación con el sexo masculino se esperan 2.700 nuevos diagnósticos de cáncer en el 2019, correspondiendo el 29,1% (774 casos) a cáncer de próstata, siendo la mayor morbilidad por cáncer en ese periodo.7
Diferentes estudios han identificado tanto alteraciones genéticas como epigenéticas en los cánceres antes listados. Sin embargo, el papel de las alteraciones epigenéticas es interesante porque, a diferencia de otros cambios genéticos (mutaciones) estas alteraciones son reversibles, por lo que particularmente pueden ser susceptibles a las terapias farmacológicas. Luego, para comprender el impacto de los procesos epigenéticos en cáncer, es necesario reconocer las alteraciones epigenéticas más frecuentes que se presentan en los cánceres más prevalentes en nuestra región, que permitan una mejor comprensión del proceso de transformación celular y tisular, y por lo tanto un mejor manejo del paciente en los momentos de diagnóstico, pronóstico e intervención terapéutica.
Además de proporcionar nuevos blancos farmacológicos que podrían contribuir a la mejoría de la sobrevida de los pacientes que padecen este tipo de patologías crónicas. Finalmente, y entendiendo que el cáncer es una patología multifactorial y que el conocimiento sobre las alteraciones epigenéticas permitirá abrir nuevos espacios para la investigación biomédica sobre todo en el campo de la oncología. A través de esta revisión se pretende determinar las principales alteraciones epigenéticas asociadas al desarrollo del cáncer de próstata, mama, colorrectal y gástrico, descritas en la literatura científica durante el periodo entre enero de 2004 a mayo de 2019.
Materiales y métodos
Se llevó a cabo una revisión de la literatura científica en la base de datos Medline y Cochrane Library. Se incluyeron artículos publicados entre el periodo de Enero del 2004 a Mayo de 2019.
Se buscaron los términos MeSH/DeCS «Epigenomics», «ncRNA», «DNA Hypermethylation», «DNA Hypomethylation» «Colonic neoplasms», «Histone modification», «Breast neoplasms», «Stomach neoplasms» y «Prostatic neoplasms» en el apartado de título y/o resumen.
Los resultados se limitaron a incluir sólo hallazgos que involucran a seres humanos, en idioma inglés o español; nos restringimos a artículos disponibles de manera gratuita o a los que nos permitía acceso mediante la afiliación institucional. Se revisó la bibliografía de los artículos encontrados con objeto de hacer seguimiento de las referencias. Después de la lectura del abstract, los documentos se examinaron para identificar los potencialmente relevantes.
Estos, fueron elegibles para su inclusión si cumplían con los siguientes criterios: Estudio que relaciona alteraciones epigenéticas con el desarrollo de cáncer de cólon y/o mama y/o próstata y/o estómago; y de revisión que incluyan cáncer de mama y/o cólon y/o estómago y/o próstata. Se excluyeron los comentarios, editoriales, y cartas al editor, igualmente los publicados fuera del periodo establecido y en especies distintas a humanos.
Mecanismos Epigenéticos y su asociación con el desarrollo de cáncer
Las células somáticas de un organismo multicelular como el humano básicamente presentan la misma información genética. Sin embargo, cada subtipo celular presenta una estructura y función específica en el organismo, debido a la expresión diferencial de nuestro genoma que es regulada mediante modificaciones epigenéticas de la cromatina. Para un mejor entendimiento de estas modificaciones, resulta preciso entender el término epigenoma: el cual se define como el conjunto de etiquetas o marcas químicas (proteínas o ácidos nucléicos) que regulan la expresión génica, sin alterar la secuencia de DNA.8 De hecho, existen una gran cantidad de marcas epigenéticas que retratan la diversidad del espacio epigenético y la complejidad de la regulación génica.
Estas marcas epigenéticas se pueden clasificar como: 1. Writers, que ubican varias modificaciones químicas a un sitio específico del DNA o cola de las histonas. 2. Readers, que identifican e interpretan la señal química de acuerdo al tipo de modificación, y 3. Ereasers, que remueven la marca epigenética.9 Actualmente se conoce que estas marcas epigenéticas están asociadas con una gran variedad de enfermedades como el cáncer.
Ahora bien, dependiendo del tipo de marcador epigenético, las modificaciones en la cromatina permitirán la inhibición o activación de la expresión génica. A continuación una breve descripción de los tres principales mecanismos epigenéticos (Figura 1).
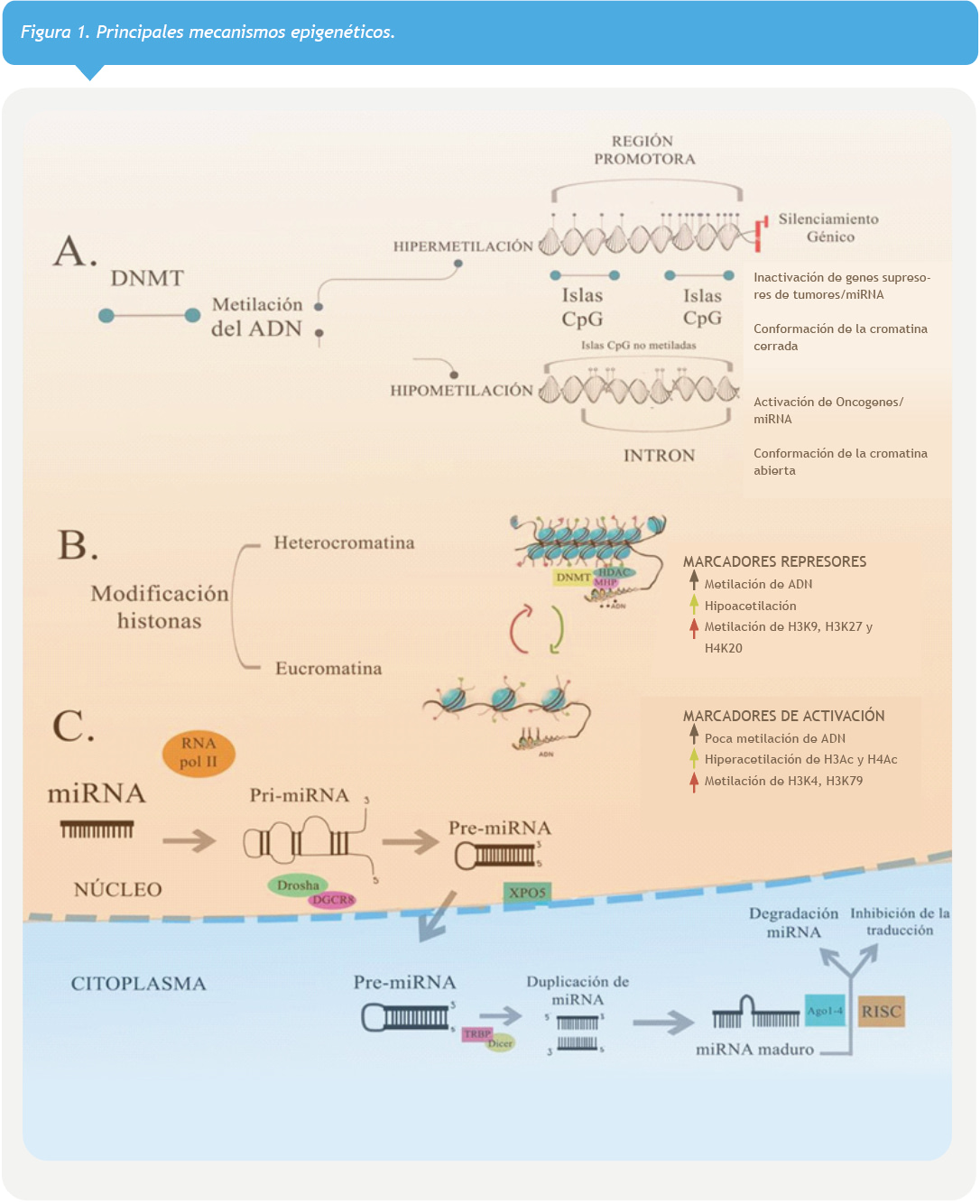
Metilación de DNA
Este mecanismo es realizado por las metiltransferasas del DNA. Su función es agregar un grupo metilo al carbono de la posición 5 en las citosinas en el DNA. Tal evento ocurre principalmente en las islas CpG (conformadas por dinucleótidos de Citosina y Guanina unidos por un fosfato, localizadas principalmente en la región central del promotor, y el sitio de inicio de la transcripción de un gen).10 La presencia del grupo metilo conduce a la no expresión de éste gen, pues confiere una variación espacial y temporal a la estructura de la cromatina, en donde a mayor metilación, menor expresión génica y viceversa. La modificación dificulta la unión entre el gen y la maquinaria transcripcional. Este proceso es mantenido en el tiempo por la DNMT1 (Figura 1A).

Modificaciones en la cola de las Histonas
La metilación y acetilación son las principales modificaciones en la cola de las histonas. Estos procesos se logran por acción de proteínas que ubican o retiran los grupos metilo o acetilo. Para la metilación están las Histona Metiltransferasa y la Histona Desmetilasa que metilan y desmetilan respectivamente. En su contraparte se encuentran las Histona Acetiltransferasa e Histona Desacetilasa involucradas en la acetilación. Las histonas tienen una cola amino terminal que es donde se ubican las modificaciones químicas como metilación de residuos de Lisina y Arginina y acetilación de residuos de lisina.11 Entre otras modificaciones está la ubiquitinización, fosforilación y sumoilación, pero las más estudiadas son las inicialmente descritas. Las enzimas metiltransferasa y acetil desmetilasa tienen la función de generar una unión fuerte entre DNA e Histona, al igual que de histona a histona, de esta manera, se genera el silenciamiento de genes.
Por el contrario, la Acetiltransferasa y la Desmetilasa funcionan aliviando la tensión entre ambas hebras y unión entre histonas, favoreciendo la lectura y transcripción de los genes (Figura 1B).
RNAs no codificantes
Son pequeñas moléculas de RNA con capacidad de unirse a RNAs más grandes e impedir su funcionamiento o la síntesis de proteínas. Son de gran importancia en procesos de generación de proteínas y en el silenciamiento de los genes. Dentro de los RNAs no codificantes se pueden mencionar a los RNA tipo ribosomas que cumplen la función de generar la síntesis de proteínas, los RNA de los empalmosomas, encargados del corte y empalme de pre RNAm; los miRNA que tiene la función de unirse a una secuencia de DNA y generar silenciamiento, al igual que regular la conformación de la cromatina, pueden inducir la transcripción al unirse a los factores de transcripción adecuados, pueden inhibir la transcripción de genes, entre otras(Figura 1C).3 En la actualidad se han acumulado un número importante de estudios donde se observan alteraciones epigenéticas que participan en los diferentes estadios del cáncer, desde su inicio en células premalignas o durante las progresión del tumor, participando no solo en la severidad sino también en su grado de malignidad o metástasis. Y aunque en algunos casos los datos no son muy precisos, las observaciones obtenidas en diferentes estudios realizados en lesiones pre-malignas, tumores primarios y modelos experimentales, han permitido el diseño de estrategias terapéuticas encaminadas a revertir estas alteraciones en pro del manejo y pronóstico de los pacientes con cáncer. Por lo anterior, a continuación se presentan los cambios epigenéticos más frecuentemente observados en los cuatro tipos de cáncer más prevalentes a nivel mundial y regional.

Cambios Epigenéticos en Cáncer Colorrectal (Crc)
Al cáncer colorrectal se atribuyen 4.489 muertes en Colombia durante el 2018 siendo la tercera causa de muerte por neoplasias. 12 Se destaca la divergencia de ambos sexos en incidencia y mortalidad, siendo de predominio femenino para ambos casos.
Metilación del DNA en CRC
Feinberg y Vogelstein fueron los primeros en observar que ocurrían variaciones en la metilación del DNA en los tejidos cancerígenos del cólon.13 Luego de este descubrimiento se han ido identificando cada vez más genes metilados anormalmente.
Este proceso influye en distintas vías de señalización como la WNT con los genes APC y AXIN2; vías de reparación del DNA como miss-match con reparación de mal apareamiento de bases, y la vía de O-6 metilguanina metiltransferasa con genes MGMT, MLH1 y MLH2; y genes implicados en el ciclo celular como CDKN2A, entre otros.14

A través de estudios de amplificación de islas CpG metiladas, se demostró que hay un subconjunto de CRC que tienen patrones distintivos en la metilación de dichas islas, ello se denominó “Fenotipo de Metilación de Islas CpG” o CIMP. Se caracterizan por presentar alto porcentaje de promotores de genes metilados de forma aberrante.15 El status del CIMP permite clasificar los CRC en 3 subgrupos: CIMP1, CIMP2 y CIMP-negativo. Cada uno de los estatus de CIMP se correlaciona con la presencia o ausencia de MSI, mutaciones en BRAF, K-ras y/o p53. El CIMP1 tiene MSI y mutaciones en BRAF; CIMP2 tiene mutaciones en K-ras; y CIMP-negativo tiene mutaciones en p53 sin mutar BRAF ni K-ras, e igualmente carece de MSI.16
El mecanismo mediante el cual interactúan el estatus del CIMP con las mutaciones en K-ras/BRAF se basa en que la vía de Ras que activa proteínas que median el silenciamiento génico como la DNMT1 a través de metilación, modificando el CIMP; del mismo modo los genes afectados por el CIMP parecen estar involucrados en la transformación celular mediada por la activación de la vía de K-ras/BRAF, que induce a la senescencia celular. Por tal razón se favorece la génesis tumoral, pues los genes que inducen a senescencia a través del Ras son metilados, impidiendo su función; así pues los tejidos de CRC evaden la senescencia mediante la inactivación de sus inductores (p16) y mediante activación de señales oncogénicas (mutaciones en BRAF).16
La progresión a malignidad se ha considerado un proceso que implica acumulación de mutaciones en el tiempo asociado a alteraciones epigenéticas que cambian la producción de proteínas y el curso del tejido, estas alteraciones pueden darse tempranamente en el desarrollo del tumor como ocurre con la metilación del gen MGMT, que codifica para una proteína que actúa como enzima reparadora del DNA removiendo grupos alquilo de las guaninas; o de forma tardía, como es el caso de la hipermetilación del gen MLH1 que se encuentra en el 15% de los CRC esporádicos y se reconoce como un evento tardío en la progresión del Adenoma serrado y su transición a cáncer colorrectal.14 RUNX3 es un gen supresor de tumores que participa en el funcionamiento de células de la respuesta inmune innata y adaptativa; se ha relacionado con patologías inmunitarias como la enfermedad de Crohn y colitis ulcerativa así como cáncer gástrico y colorrectal. Este control inmunitario e inflamatorio ejercido por RUNX3 influencia el desarrollo de tumores epiteliales.17 Su inactivación se debe a hipermetilación de la región promotora. La hipometilación también se ha descrito en la patogénesis del CRC, encontrándose hipometilación global del DNA, se cree que participa en el desarrollo de la enfermedad induciendo inestabilidad cromosomal y pérdida global de imprinting.18
Modificaciones en la cola de las histonas en CRC
La marca epigenética más estable es la metilación del DNA, las modificaciones en la cola de las histonas son biomarcadores más inestables y por ello su investigación ha sido limitada.14 Se ha encontrado hipoacetilación global en las lisinas de histonas H3 y H4 en líneas celulares de CRC, y como consecuencia hay pobre regulación de genes supresores de tumores y genes supresores de metástasis.14 La sobre-expresión de la Acetiltransferasa de Histonas en CBP, aparece en estadios tumorales avanzados y se relaciona con crecimiento celular y metástasis, esto dado por la acetilación de MTA2 que pertenece a la familia de las Proteínas asociadas a metástasis. A su vez, la acción aumentada de las HDAC clase 1 a 3 se relaciona a pobre regulación de genes pertenecientes a la vía de señalización de Wnt que participa en proliferación celular.19

Hay pérdida de la trimetilación de H3K20, concomitante a dimetilación y trimetilación de H3K4, H3K9 y H3K27. Tales variaciones en la metilación de histonas repercuten en características propias del tumor como tamaño, tipo histológico, tendencia a la invasión de nodos linfáticos, y metástasis.14 Puntualmente, la metilación constante dada por PRC1 sobre H3K27 se correlaciona con bajas tasas de supervivencia. Adicionalmente, las desmetilasas de histonas tipo KDM4 están sobre-expresadas en tejidos de CRC y gracias a su capacidad de modular factores de transcripción contribuyen al crecimiento celular en este tipo de tejidos.14
El UHRF1 es un factor epigenético que regula la transcripción modulando la metilación del DNA y la modificación de histonas.
Ejerce efectos sobre el gen supresor de tumores TUSC3 al situarse sobre la región promotora de este último, resultando en regulación negativa de la acetiltransferasa de histonas KAT7. Lo anterior resulta en menor acetilación del TUSC3, favoreciendo la proliferación celular en cáncer de cólon.20
RNAs no codificantes en CRC
Los miRNAs y los lncRNA son los RNA no codificantes más observados en CRC. Al igual que la metilación del DNA, estos RNA’s cumplen funciones en relación a la progresión tumoral de acuerdo a la fase en que se encuentre el tejido, se expresan algunos tempranamente como miR-135b y miR-143 que están involucrados en la iniciación del CRC por su participación en la vía de señalización WNT/β-Catenina. Mientras que otros aparecen en el proceso de transición de adenoma temprano hacia avanzado, como ocurre con miR-21 y miR-1, regulando la función de RAS-MAPK y PI3K/AKT respectivamente. Por su parte, el gen supresor de tumores p53 es regulado por el RNA no codificante miR-133a en el paso de adenoma avanzado a adenocarcinoma.18
Una nueva clase de ncRNA denominada piRNAs participa en la regulación de la metilación del DNA, silenciamiento de transposones y modificaciones en la cromatina. Se ha comprobado la expresión de proteínas PIWI de forma selectiva en células madre precancerosas, tejidos malignos y líneas celulares tumorales.21 Se involucran en el crecimiento de cáncer de colon, gástrico, renal, endometrial, ovárico, mama y pulmón.22
Cambios Epigenéticos en Cáncer de Próstata
Siendo el segundo cáncer de mayor incidencia en hombres en el mundo, y el primero en Estados Unidos para el 2016 (101,4 casos por cada 100.000 habitantes),23 resulta importante tener en cuenta su impacto dentro del territorio nacional. Según el estudio realizado por la Universidad del Valle, en su revista Colombia Médica, entre los años 2017 y 2018 la incidencia del cáncer de próstata en Colombia fue de 46,5 casos por cada 100.000 habitantes, mientras que para la capital del Valle este número aumentó hasta 59,7 casos por cada 100.000 habitantes.24 Panorama preocupante, pero poco comprendido. Dada su importancia se procede a mencionar algunas de las alteraciones epigenéticas que promueven (más no generan por sí solos) la aparición de este tipo de cáncer.
Metilación del DNA en cáncer de próstata
Uno de los eventos que más se repiten dentro de las etapas iniciales de aparición del cáncer de próstata es la hipermetilación de la región promotora del gen GSTP1, familia enzimática encargada de la desintoxicación celular por medio de catalizar la conjugación de compuestos electrofílicos e hidrofóbicos que contiene una carga de glutatión reducida, protegiendo a la célula del daño oxidativo producto de compuestos carcinogénicos del ambiente.25 De acuerdo a las investigaciones de Lee et al,26 “90% de los carcinomas prostáticos presentan silenciamiento del gen GSTP1 debido a la hipermetilación de la CpG 5”. Alteraciones en la región promotora del gen que codifica para el AR se han relacionado con la aparición de cáncer de próstata no dependiente de andrógenos, pues se ha visto que la hipermetilación del DNA en tales regiones promotoras del gen contribuye a la independencia del tumor frente a los andrógenos. Paradójicamente entre el 20% y el 30% de los cáncer de próstata no dependientes de andrógenos cuentan con una expresión normal del gen.27
Históricamente se han utilizado terapias hormonales con estrógenos para enlentecer el proceso de crecimiento y proliferación de los tumores prostáticos. Originalmente se creía que bloqueaban el eje Hipófisis-gonadal, impidiendo la producción de andrógenos, pero gracias a investigaciones recientes, se cree que sus efectos terapéuticos se deben a su acción directa sobre los ESR, específicamente en ESR1 (ERalfa) y ESR2 (ERbeta). Lo anterior explica por qué los pacientes con hipermetilación en las islas CpG de la región promotora para ESR2 presentan un peor pronóstico, ya que no expresan receptores para estrógenos en la superficie de las células epiteliales prostáticas. En adición se ha visto que los ERbeta tienen un efecto antiinflamatorio, antioxidante y antiproliferativo, acciones que son contrastadas por su homólogo ERalfa, el cual se encuentra en células estromales y está involucrado en actividades de proliferación y crecimiento prostático.
Específicamente en cáncer de próstata, la expresión de ERalfa se encuentra silenciado en etapas tempranas y de manera paulatina va aumentando de acuerdo con la progresión de este cáncer, por su lado el ERbeta tiene un efecto paradójico, en etapas tempranas debe tener una baja expresión para permitir la progresión a neoplasia y aumenta de manera significativa antes y durante el paso a metástasis. Lo que en un futuro, y con mayores estudios podría darnos información de utilidad para su uso como indicador del estado del tumor y nos ayudaría a generar mejores tratamientos.28
Las CDKI son una superfamilia de proteínas reguladoras del ciclo celular, encargadas de detenerlo al detectar errores en el proceso de apareamiento de bases, o daño en la molécula de DNA. Dentro de esta superfamilia se encuentra la CDKN2A. El fallo en alguna de las ciclinas de este grupo representa un factor de alto impacto para la aparición de varios tipos de cáncer. Curiosamente la inactivación del gen de CDKN2A mediada por la metilación del exón 2 se encuentra en el 66% de los cánceres de próstata, a diferencia de su silenciamiento por hipermetilación de su región promotora que ocurre en casos aislados. Debido a que el silenciamiento de este gen sólo ocurre en el cáncer de próstata, y es un evento raro que otras ciclinas se vean involucradas en su aparición, la utilización de este fenómeno como un biomarcador para la detección temprana de este cáncer resulta de gran ayuda, dado que se ha postulado que al ocurrir la metilaciones de promotores de este gen, ocurre la detonación para el inicio del evento neoplásico.29
Estudios experimentales reportan que la hipermetilación del exón 2 no genera impedimento para el funcionamiento correcto de la CDKN2A, el mecanismo por el cual se da la inactivación de este gen en tejidos vivos extraídos y líneas celulares, no está bien dilucidado.27
La familia de proteínas denominada Moléculas de Adhesión Celular, es indispensable para generar el sistema Cadherina- Catenina, que permite la fijación de células en tejidos específicos.
Dicho sistema es importante para regular procesos de renovación, crecimiento y proliferación celular. Son moléculas clave para la identificación de la progresión de múltiples cánceres.30 La CDH1, conocida como E-cadherina, es un marcador molecular importante para detectar la progresión tumoral. En el 30% de los tumores de bajo grado y 70% de los de alto grado se ha encontrado 28 hipermetilaciones en los CpG cercanos al promotor, así como metilación gradual del primer exón del gen CDH1; metilación que aumenta de acuerdo al grado de progresión del tumor, demostrando una caída constante de la síntesis de E-cadherina hasta llegar a estadíos metastásicos donde esta proteína es ausente.27
Se ha visto que la cantidad de hipermetilaciones dentro del genoma es mínima a comparación de la disminución de la 5-metilcitosina, condición que se ha visto en múltiples cánceres en etapa inicial.31 El mecanismo por el cual la hipometilación se asocia a cáncer no está bien dilucidado, hasta ahora se ha encontrado que fallos en la función de la DNMT1 corresponden con el aumento de la hipometilación, la cual contribuye a la disminución del crecimiento tumoral en algunos modelos murinos. De manera contradictoria contribuye al crecimiento tumoral en otros modelos.
Lo que si se tiene claro es que la metilación por 5-metilcitosina se encuentra disminuida en aquellas células prostáticas que presentan hiperplasia o procesos metastásicos.32
Generalmente las células cancerosas, a diferencia de las normales, se encuentran altamente hipometiladas, lo que contribuye a la sobre expresión de ciertos genes que promueven el crecimiento celular, la supervivencia y la migración. Uno de ese caso es el del gen PLAU el cual en condiciones normales permanece con su locus transcripcional metilado. Cuando no se encuentra en este estado, codifica para una proteína multifuncional que en el caso del cáncer de próstata promueve la invasión y metástasis tumoral, al igual que en desarrollo de cáncer prostático complejo independiente de hormonas.33
Modificación de histonas en Cáncer de Próstata
La aparición del cáncer de próstata se ha visto relacionada con la activación o silenciamiento de diversos genes dependientes de la modificación de histonas. Entre estos se encuentran genes como el CXADR, el cual es indispensable por los Adenovirus tipo C para su invasión celular. En cáncer se ha visto que cuando hay una acetilación de histonas que comprometen la región que contiene este gen, se induce la aparición de líneas celulares cancerígenas tipo PC-3 (carcinoma prostático de células pequeñas).27 Otro gen que se ha visto afectado por la modificación de histonas, es el gen VDR, codificante para receptor de vitamina D, esta última siendo importante para permitir la entrada de 1,25 hidroxi vitamina D al ciclo celular y así ejercer un efecto antiproliferativo de las células tumorales.
Por medio de la acetilación de histonas, se ha visto que hay silenciamiento de este receptor y por ende imposibilidad de regulación del ciclo celular mediada por la vitamina D. Coincidencialmente se ha visto que hay un subtipo de células tumorales prostáticas, que no presentan esta alteración en las histonas que se encuentran sobre el gen para el receptor de Vitamina D, en las cuales a pesar de tener este tipo de receptores, desarrolla un mecanismo de defensa secundario el cual consiste en la sobre expresión de receptores nucleares tipo SMRT el cual se encarga de aumentar la actividad deacetilasa sobre las histonas en sinergia a la disminución de la actividad transcripcional para el gen del receptor de vitamina D.34
La metilación de la histona 3 en su lisina 4 se ha asociado con la inactivación de la transcripción del gen que codifica para la molécula del antígeno prostático (PSA), antígeno útil como prueba de tamizaje, en conjunto al examen físico, en adultos entre los 55 y 69 años o en aquellos con historial familiar, al igual que ser útil como estimador de la tasa de supervivencias post diagnóstico, como indicador de respuesta a la terapia y en conjunto con la muestra histológica importante para medir probabilidad de recurrencia. Cuando esta molécula no se encuentra presente, lleva a la aparición de tumores de próstata de líneas celulares LNCaP, las cuales, al ser indetectables por laboratorio en sus fases iniciales, generan un diagnóstico más tardío de la enfermedad.35
También se han visto en los cánceres resistentes a radioterapia la hipoactividad o ausencia completa de la actividad de HDAC.36
Interacción entre Metilación de DNA y modificación de histonas
Si bien cada evento por separado puede desencadenar en desarrollo de células malignas, al juntarse los dos procesos conllevan a la aparición de un estado denominado de inactividad transcripcional de la cromatina, en donde por medio de la unión de proteínas de adhesión a DNA metilado, como en el caso de la MeCP2, se promueve el reclutamiento de HDAC el cual a su vez metila los promotores del gen afectado llevando al silenciamiento de este.
Adicionalmente otra molécula como las DNMTs pueden reclutar directamente a las HDAC para silenciar la expresión de genes.31 In vitro se ha comprobado que otro gen afectado por este efecto doble es el gen RARB, el cual se ha encontrado metilado en la mayoría de los cánceres de próstata, a su vez que ocurre una hipoacetilacion en las histonas H3 y H4 en las líneas celulares de este cáncer.
Aunque aún no se ha dilucidado cuál de los dos eventos ocurre primero, en el cáncer de próstata con células LNCaP, se ha visto que primero debe ocurrir la metilación directa del gen GSTP1, el cual es seguido por la acetilación de histonas.27
Cambios Epigenéticos en Cáncer de Mama
En las últimas décadas se ha visto un incremento significativo en la incidencia del cáncer de mama la cual es muy variable en los diferentes países a nivel mundial, esto probablemente relacionado con el aumento de la esperanza de vida y los factores ambientales a los que la población se enfrenta. Este tipo de cáncer representa un 16% del total de los cánceres femeninos, con una supervivencia relativa a 5 años al igual que el cáncer colorrectal y de próstata.37,38 La prevalencia de cáncer de mama es más frecuente en mujeres que en hombres, se estima que en los Estados Unidos para el año 2019 se van a reportar alrededor de 268.800 casos nuevos en pacientes de sexo femenino y cerca de 2.670 casos en hombres.39 No existe una clara diferencia entre países en vías de desarrollo y países desarrollados, aunque se ha encontrado una mayor mortalidad en países en vía de desarrollo. Los principales tipos histológicos que se han descrito son: el carcinoma de mama infiltrante con una prevalencia cercana al 76%, y otros con una prevalencia menor como el lobular invasivo (8%), y ductal/lobular (7%).40
Metilación del DNA en cáncer de mama
Los cambios epigenéticos que afectan la familia de las metiltransferasa presentan una relación directa con respecto a los cambios epigenéticos que conllevan a un proceso de carcinogénesis.
Hasta el momento se han descrito más de 8 tipos de estas metil transferasas, las que presentan actividad catalítica son: DNMT1, DNMT3A y DNMT3B. Se ha demostrado que la metilación diferencial en cáncer de mama en comparación con tejidos sanos, manifiesta un impacto en el proceso de salud y enfermedad que puede ser indicador o no de metástasis. Diferentes estudios han mostrado niveles aumentados en la expresión de ciertos genes relacionados con esta familia, tal es el caso del DNMT3b que se encuentra en niveles más altos en células tumorales comparada con células normales.41-43
El proceso de carcinogénesis se ha asociado a la metilación en la región promotora de algunos genes como BRCA1, MLH1, y MGMT.44 Existe evidencia sólida que relaciona a la hipermetilación del promotor de BRCA1 con carcinoma de mama, su incidencia varía según el estudio y oscila entre un rango aproximado de un 7-30% en pacientes con cáncer de mama esporádico. BRCA1 es un gen supresor de tumores, a su vez se encuentra involucrado en el mantenimiento y reparación del ADN, la metilación en el promotor de este gen puede desencadenar un proceso tumoral mediante 2 vías, en primer lugar la hereditaria, generada por mutaciones propias del gen y en segundo lugar las alteraciones epigenéticas. El silenciamiento de BRCA1 incrementa múltiples alteraciones que incluyen mutaciones en genes “passengers”.
Estas alteraciones epigenéticas son más prevalentes en cáncer de mama ER, PR y HER2 negativos.44,45
En otro estudio tomaron muestras de pacientes menores de 50 años en donde encontraron que un 14,8% presentaba alteraciones de BRCA1, mientras que en las mujeres mayores a los 50 años solo un 3% mostró cambios epigenéticos en los promotores de este gen, lo que sugiere que la hipermetilación de BRCA1 tiene mayor prevalencia en mujeres premenopáusicas y perimenopáusicas, comparado con las post-menopáusicas.45
Por otra parte varios estudios han correlacionado la hipermetilación de INK4A con carcinoma de mama y colorrectal, el silenciamiento de GSTP1 se ha asociado a cáncer de mama y próstata. Éste mismo mecanismo epigenético se ha dilucidado en los promotores de algunos genes de reparación del ADN como el gen MGMT, el cual se ha asociado tanto en cáncer de mama como colorrectal. Cabe mencionar que estos procesos se pueden relacionar con el estadio metastásico del tumor, el silenciamiento de CDH1 se observa en la mayoría de los pacientes con cáncer de seno metastásico, por lo cual encontrar el promotor del gen CDH1 hipermetilado supone un peor pronóstico para el paciente.43,46 Diferentes estudios han descrito una frecuencia alta de alteraciones epigenéticas relacionadas con genes antagonistas de la señalización de Wnt, su metilación se asocia a procesos tumorales.
Así como la anterior, existen alteraciones epigenéticas que se correlacionan con la progresión del proceso tumoral, tal es el caso del silenciamiento epigenético mediante hipermetilación de NEFH y NELF. El ensamble que ocurre entre NEFH y NELF permite la formación de neurofilamentos, por lo que su inactivación conduce a una desregulación proteica. Estudios han demostrado que al recuperar la expresión de NEFH se correlaciona directamente con inhibición de la proliferación de las células tumorales.43,47
La hiper o hipometilación de RASSF1A y CCND2 se correlaciona con la progresión del cáncer de mama, asociada principalmente a cánceres ER+, por otro lado en el cancer de mama ER- se encontró hipermetilación de PGR, TFF1, CDH13, TIMP3, HSD17B4, ESR1 y BCL2.43,48 Otros estudios han encontrado en carcinoma de mama ER – los genes RIL y CDH13 metilados.44 Algunos estudios han encontrado diferencias entre pacientes afroamericanas y caucásicas en donde mujeres menores a los 50 años caucásicas presentaban menor metilación de HIN-1, TWIST1, CCND2 y RASSF1A en comparación con las afroamericanas.49
Los estudios de carcinoma de mama y su correlación con el estadio del receptor hormonal han ido tomando relevancia en los últimos años ya que está relacionado al pronóstico y a la respuesta farmacológica a medicamentos como tamoxifeno y los inhibidores de la aromatasa. Un estudio publicado por Lian li48 realizó una comparación entre los estados de metilación en cáncer de mama con ER+ / PR+ y 12 ER− / PR− , se encontró una diferencia en cuanto a la respuesta a la terapia hormonal en donde los tumores ER+ /PR+ responden mejor a terapia hormonal a su vez estos también se han asociado a la menarquia temprana y la obesidad posmenopáusica.48 Existen otros estudios que no se relacionan directamente con el estadio hormonal del tumor, sin embargo también pueden tener un valor pronóstico importante y a su vez correlacionarse con la progresión y crecimiento del tumor. Un ejemplo de esto es el patrón de la expresión del gen CXCR4 con su ligando CXCL12, en donde la no metilación de CXCR4 se ha asociado con el estadio, tamaño, y el grado histológico del tumor y con el aumento en la probabilidad de la metástasis y mortalidad.50
Por otro lado, FEN1 es una nucleasa que participa en la replicación del DNA permitiendo la maduración de los fragmentos de okasaki, en caso de ser estimulada participa en la fragmentación del DNA inducido por apoptosis, mantiene la estabilidad genómica, su déficit se relaciona con una mayor velocidad en la formación de tumores y predisposición al cáncer. La hipometilación de este gen se encontró aumentada alrededor de 2,5 veces más en muestras de tejido tumoral, con tendencia a presentar una mayor prevalencia en carcinoma lobular infiltrante de mama. Por otro lado es importante describir que también se ha encontrado aumentada hasta dos veces en cáncer colorrectal y gástrico. Su hipometilación se encuentra relacionada con el pronóstico y la capacidad de realizar metástasis.51
El silenciamiento de la vía TGF- β producto de cambios en la remodelación de la cromatina a partir de cambios en la metilación de la región promotora del gen SNFE1/SWI se han relacionado
con una sobreexpresión de la MYC relacionándolo por lo tanto con procesos como metástasis no obstante, durante el inicio de la carcinogénesis este es parte fundamental de la transición epitelio-mesenquimatosa. A su vez cambios en algunos genes de la señalización de las integrinas también se han visto afectados, tanto su hiper o hipometilación se han correlacionado con carcinoma de mama. En el caso de ZEB2 y SNAIL2 también pueden estar hiper o hipometilados dependiendo de la fase de progresión del tumor, cuando se encuentran hipometilados se han correlacionado con la inhibición de CDH1 y por lo tanto un aumento en la metástasis.41,43
miRNA en cáncer de mama
La expresión de los miRNA se correlacionan con el estadio del tumor, y pueden relacionarse con un aumento de las mutaciones por ejemplo miR-210 asociado a alteraciones en BRCA1 y Ecadherina.
Se ha demostrado diferencias importantes al comparar muestras de tejidos de cáncer de mama con células epiteliales mamarias humanas normales. Un estudio realizado por Lukas Vrba y sus colaboradores encontraron que un tercio de las muestras obtenidas en cáncer de mama mostraron una metilación aberrante del ADN en los promotores de mir-31, mir-130a, let-7a-3 / let-7b, mir-155, mir-137 y mir-34b / mir-34c lo cual se ha relacionado con una desregulación del miRNA y se ha asociado a un aumento del potencial de metástasis en células cancerígenas. Por otro lado la baja expresión de miR-12b, miR-145, miR-21 y miR-155 se ha descrito en diferentes tipos de cáncer de mama. Otras investigaciones encontraron la baja regulación de los miRNA let-7d, miR-210, miR-221 en el carcinoma ductal in-situ.41,52
Modificaciones de histonas en cáncer de mama
La familia de las proteínas MBP está compuesta por MeCP2, MBD1, MBD2, MBD3 y MBD4. Al igual que las DNMT, median el silenciamiento de genes supresores de tumores a través de la hipermetilación de las islas CpG y la interacción entre ADN y enzimas modificadoras de histonas. La HMT y las HDAC también se han correlacionado con el silenciamiento y la inactivación de genes supresores de tumores, es relevante mencionar que este proceso requiere de la interacción con las DNMT.43
En carcinoma de mama basal triple negativo y los HER2+ las modificaciones de histonas juegan un papel fundamental ya que se ha encontrado que tanto la baja acetilación como la metilación de la lisina y arginina se correlacionan con un peor pronóstico.41
La disminución leve de la acetilación de las histonas ac-H4, acH4K12, ac-tubulina, HDAC1, HDAC2 y HDAC6 se ha visto con mayor frecuencia en carcinoma ductal mientras que trimetilacion de K20 de la histona 4 o la pérdida generalizada de la acetilación en la K16 se han encontrado cáncer gástrico y de próstata. En carcinoma de mama tipo luminal entre menor sea el grado de acetilación se considera que el paciente presenta un mejor pronóstico.53
Cambios Epigenéticos en Cáncer Gástrico
En Centroamérica y Sudamérica se encuentra el 17% de los cánceres gástricos del mundo, con una incidencia 2 a 3 veces mayor en hombres que en mujeres. En Cali (Colombia) según el registro poblacional de Cáncer, el carcinoma gástrico ocupa el cuarto puesto de incidencia en ambos sexos, siendo 8,1% similar a la de cáncer de colon y recto.38 Además el atlas de mortalidad de cáncer del país reveló que el cáncer gástrico es la primera causa de mortalidad por cáncer en hombres y la tercera en mujeres representando un 13,7% de todas las muertes por cáncer en el país.39
Modificación del DNA en cáncer gástrico
En el cáncer gástrico se han observado distintas alteraciones epigenéticas, entre estas encontramos que la hipermetilación del promotor del gen reparador MLH1 es uno de los principales mecanismos responsables del cáncer gástrico, junto con la hipermetilación de p16 en el subtipo intestinal. Los genes de p16, p14 y CDKN2B/p15 se encuentran involucrados en tumores primarios en líneas celulares humanas, el p16 es silenciado por la hipermetilación en su región promotora y se ha asociado al cáncer gástrico, siendo así que puede predecir la progresión a malignidad en displasias.56 La metilación de CDKN2A en su promotor se asocia a progresión de lesiones precursoras hacia malignidad Metilación del promotor de MLH1 es frecuente en la mayoría de cáncer gástricos y en un 71% de los que presenta inestabilidad por microsatélites. El 31% de las hipermetilaciones del reparador de MGMT también se han asociado con cáncer gástrico.57
La capacidad de invadir y migrar a otros tejidos se ha relacionado con la hipermetilación del promotor de CDH1 encontrada en cánceres difusos y la alteración en la regulación de E-cadherina se ha asociado con pobre pronóstico.58 El CDH4 se encuentra hipermetilado en varios cánceres gástricos. La hipermetilación de DAPK se observa en el subtipo difuso o en el mixto y en casos de metástasis. La inactivación de RAS, especialmente al dominio familiar uno isoforma A (RASSF1A) mediante metilación está presente en cánceres de mama, pulmón y cáncer gástrico primario.57
El XIAP es una proteína antiapoptótica que actúa sobre las caspasas. El silenciamiento del Factor XAF1 por metilación aberrante se encuentra en el cáncer gástrico. En cánceres primarios la hipermetilación de TSPY L5 en regiones CpG ocurre en 23 de cada 36 tumores.57 El hSRBC incrementa la estabilidad de P53 y la expresión de sus genes blanco, la pérdida de hSRBC se ha encontrado en 41% de los cánceres gástricos primarios. La expresión de SFRP2 se relaciona con la hipermetilación del promotor en 73,3% de los casos y la metilación de SFRP1, SFPR5 y SFRP2 en ambas líneas celulares de cáncer gástrico primario.57 La metilación de DKK también se ha reportado en líneas celulares de cáncer gástrico primario. Miembros de la superfamilia SWi/SNF pueden actuar como supresores tumorales, pero la metilación de HLTF un homólogo de estas se presenta en casi la mitad de los carcinomas gástricos. La familia RUNX en especial RUNX3 se relaciona con carcinogénesis gástrica, se encuentra metilado en 8% de las gastritis crónicas, 28% de las metaplasias intestinales, y 27% de los adenomas gástricos. La hipermetilación de TSP1 se observa en 33% de los cánceres gástricos y hepáticos.57
La gastritis de alto riesgo en infección por H. pylori se relaciona a un patrón de hipometilación global que se asocia a inestabilidad genómica. Un 70% de los cánceres gástricos están asociados a la infección por H. pylori. El proceso, aunque no bien descrito, se sospecha que comienza con gastritis crónica que conduce a atrofia glandular, metaplasia intestinal, displasia y termina en adenocarcinoma gástrico. La Hipometilación global en el DNA de las células cancerígenas fue descrito en la década de los 80s, sin asociarse a mutaciones en las DNMT.59
La hipometilación es un proceso que se acumula con la edad del paciente, el número de divisiones y se asocia con el pobre pronóstico en el cáncer gastrointestinal. En un estudio donde se midió el metil-CpG de manera global en el DNA, en tejidos tumorales frente a no tumorales se encontró que en el cáncer gástrico había significativa menor metilación. Las biopsias con H. pylori mostraron niveles reducidos de hipometilación comparado con los controles; los niveles de hipometilación permanecían aunque se erradica la bacteria. El patrón de hipometilación fue heterogéneo y ocurría principalmente en la mucosa, esto lo relacionan al número de divisiones celulares, comparando la mucosa con la serosa y la muscular.59
En un ensayo clínico de biomarcadores se busco identificar a partir de biopsias si la hipometilación global del DNA de la mucosa gástrica se relacionaba con la aparición de cáncer gástrico comparando pacientes con gastritis sin metaplasia, gastritis con metaplasia y cáncer gástrico. En este se encontró una relación inversa entre un índice de hipometilación global y el desarrollo de la neoplasia. El resultado del estudio concluyó que entre mayor fuera el índice de hipometilación (GDMI) propuesto por el estudio, el paciente sería más propenso a desarrollar cáncer o a tener la patología en curso.60
RNAs no codificantes en cáncer gástrico
Los RNA no codificantes se han visto implicados a través de diferentes estudios sobre cáncer gástrico que suelen tener como objetivo demostrar la utilidad de estos como marcadores de aparición, gravedad y pronóstico de la patología. En un ensayo clínico se ha identificado que el lncRNA ZFAS1 se encuentra sobre expresado en este tipo de cáncer, y sus altos niveles se relacionan con un peor pronóstico para pacientes con dicha neoplasia. Se identificó además que parte de su mecanismo como oncogen se basa en la represión de los genes KLF2 y NKD2 por medio del reclutamiento de las proteínas de unión a RNA PRC2 y LSD1.61 En otro estudio se encontró que el lncRNA BCO32469 se ve sobreexpresado en el tejido de cáncer gástrico, además su desregulación se vio asociada a mayor tamaño del tumor, pobre grado de diferenciación del tejido y mal pronóstico para los pacientes. Su depression in vitro e in vivo resultó en una inhibición de la proliferación del tumor. Su mecanismo oncogénico se ve asociado a su unión miR-1207-5p y provocación de la depresión de la expresión de hTERT.62 De manera similar se ha identificado que la expresión de lncRNA 00152 suprime la expresión de de p15 y p21 por medio de la unión a su promotor a través de EZH2, promoviendo así la progresión del ciclo celular de las células malignas del cáncer gástrico.63 El lncRNA HOTAIR y su sobreexpresión se ha asociado a mayor proliferación e invasión del cáncer gástrico al reprimir la expresión de genes supresores de tumores.64
Discusión
Entre los cambios epigenéticos evaluados en este artículo se encontró que estos están implicados en la expresión de genes que participan en diferentes vías entre las cuales destacan las relacionadas con reparación del DNA, apoptosis, adherencia y ciclo celular.
En el cancer de mama y de colon se encontró que la DNMT1 ejerce función de silenciamiento génico se encuentra metilada lo cual provoca la falta de metilación de otras proteínas que en últimas desregulan el ciclo celular. De igual manera las hipometilaciones de regiones inductoras de crecimiento y proliferación celular, se han visto asociadas a la aparición de los patrones cancerígenos en algunos de estos cánceres.
Estas alteraciones pueden ocurrir tanto por mutaciones que comprometan las enzimas encargadas de estos procesos o por alteraciones en su regulación ya descritos anteriormente. En cáncer de próstata los patrones de hipometilación también se encuentran descritos en la fisiopatogenia de cáncer en este caso se involucra el gen del PLAU que cuando no se encuentra apropiadamente metilado tiene repercusiones en procesos de diseminación tumoral.
La capacidad de migración de una célula tumoral se ha visto relacionada en los tres tipos de cáncer por ejemplo en el cáncer de estómago la alteración de CDH1 encontrada en cánceres difusos, la cual conlleva a un empeoramiento del pronóstico, esta misma molécula se ha visto que en cáncer de mama tiende a inhibir su expresión mediante la metilación permitiendo que estas células tumorales modifiquen sus propiedades y puedan migrar a los órganos linfoides más cercanos. Por otro lado en cáncer de estómago se ha demostrado que la hipermetilación en la región promotora de CDH1 se relaciona con alteraciones en la regulación de la E-cadherina, la cual está relacionada a infección por H. pylori.
Otra proteína de la misma familia, la CDH4, también se observa metilada en distintos tipos de cánceres gástricos y se asocia a otras mutaciones que no se encuentran presentes en cáncer de mama ni de colón, como lo es la hipermetilación de DAPK. Sin embargo, la inactivación de RAS asociada a la hipermetilación se puede observar en neoplasias de mama, pulmón y cáncer gástrico primario.
También, la metilación de RUNX3 se ha visto altamente asociado con la aparición de cáncer gástrico y colorectal.
A nivel de neoplasias del tracto gastrointestinal en este caso encontramos alteraciones en la vía de señalización de WNT/β-Catenina en tanto cáncer colorrectal como carcinoma gástrico, por lo que las vías de los RNA no codificantes son relevantes durante el proceso de desarrollo neoplásico a nivel gastrointestinal. En el cáncer de mama se ha observado también alteraciones en la regulación microRNA como miR-135b y miR-143 que comprometen la vía WNT/β-Catenina lo que indica que no es un proceso exclusivo de la carcinogénesis gastrointestinal sino que también se encuentra involucrado en otros carcinomas de diferentes tejidos. De igual manera la alteración de los RNA no codificantes como es el caso de piRNAs (Proteína PIWI de interacción RNA) es común en cáncer de mama y gástrico.
Teniendo en cuenta que el conocimiento de estas alteraciones repercuten el diagnóstico y en las propuestas de blancos terapéu ticos, el reconocimiento de hipermetilaciones de islas CpG en individuos portadores de mutaciones de alta penetrancia en genes supresores de tumor, serán útiles cuando el diagnóstico es incierto.
Por ejemplo, cuando una biopsia de mama proveniente de una portadora de una mutación en BRCA1 presenta hipermetilación de las islas CpG, esta alteración se reconoce como biomarcador temprano de desarrollo de lesiones malignas.
En relación a los tratamientos epigenéticos existen diferentes propuestas farmacológicas tales como los inhibidores de la metilación del DNA, que inhiben a las DNMTs en los casos en los que los genes supresores de tumores son silenciados. Sin embargo, la mayoría de estos inhibidores no son específicos para cada tipo de enzima y hasta el momento no se han postulado moléculas efectivas para su inhibición. Por otra parte, las moléculas LSD1 se asocian a sobreexpresión del gen ER1 que favorece el crecimiento y metástasis de las células neoplásicas. Luego si LSD1 lograra ser inhibidas o antagonizadas por otras con mayor capacidad de unión, teóricamente tendría un efecto antitumoral. Tal es el caso de cómo la molécula experimental ORY-1001, que es un inhibidor selectivo de LSD1,65 empleada inicialmente para el tratamiento de leucemia aguda, podría extender su aplicación al tratamiento de otros tipos de cáncer como los previamente discutidos.
Otro blanco importante son los miRNA involucrados en la supervivencia y angiogénesis tumoral, si se lograra bloquear su acción, específicamente sus variantes involucradas con la cascada de señal STAT3, se reduciría la complejidad al momento de tratar cánceres como el Colorrectal, en este caso, estudios in vitro con moléculas inhibidoras de STAT3, ha abierto la posibilidad de tratamiento en un futuro.66 De igual manera las alteraciones en la expresión de varios lncRNA se han estudiado en relación al cáncer gástrico mostrándose como predictores de supervivencia y diferenciación del tejido neoplásico, tal es el caso de lncRNA BCO32469, en el que su depresión mostró disminución in vitro de tejido tumoral, crecimiento tumoral en expresión de lncRNA 00152 y proliferación tumoral junto con capacidad de invasión a otros tejidos en sobreexpresión lncRNA HOTAIR.
Un blanco importante es afectar la hipermetilación de genes supresores de tumores para lo cual, se ha estudiado el uso de agentes desmetilantes en carcinoma de mama. El uso del agente desmetilante AZA para el tratamiento del carcinoma de mama podría tener efectos positivos como el mejorar la expresión de los genes supresores de tumores que han sido silenciados de forma aberrante, no obstante, existe el riesgo de que a su vez induzca la desmetilación de oncogenes, por lo tanto se podrían usar agentes citotóxicos que anulen el efecto de los oncogenes activados. La terapia combinada con inhibidores de histona deacetilasa en laboratorios mostró una inhibición sinérgica en cultivos celulares.41
Estos son algunos ejemplos de las posibles dianas terapéuticas que en años siguientes podrían facilitar la terapia de estos cánceres disminuyendo efectos adversos y posiblemente evitando el uso de quimioterapéuticos que generan daños considerables al organismo del paciente. Aún no hay terapia definitiva y recién se están dilucidando nuevos horizontes en este campo. Los mecanismos epigenéticos pueden ser revertidos, es por esto que es tan importante seguir investigando e invirtiendo en este campo, para llegar al día en que podamos tratar cada tipo de cáncer con los menores efectos adversos posibles.
Conclusiones
En el estudio se encontraron múltiples cambios epigenéticos relacionados con cada una de las neoplasias pertinentes (Figura 2).
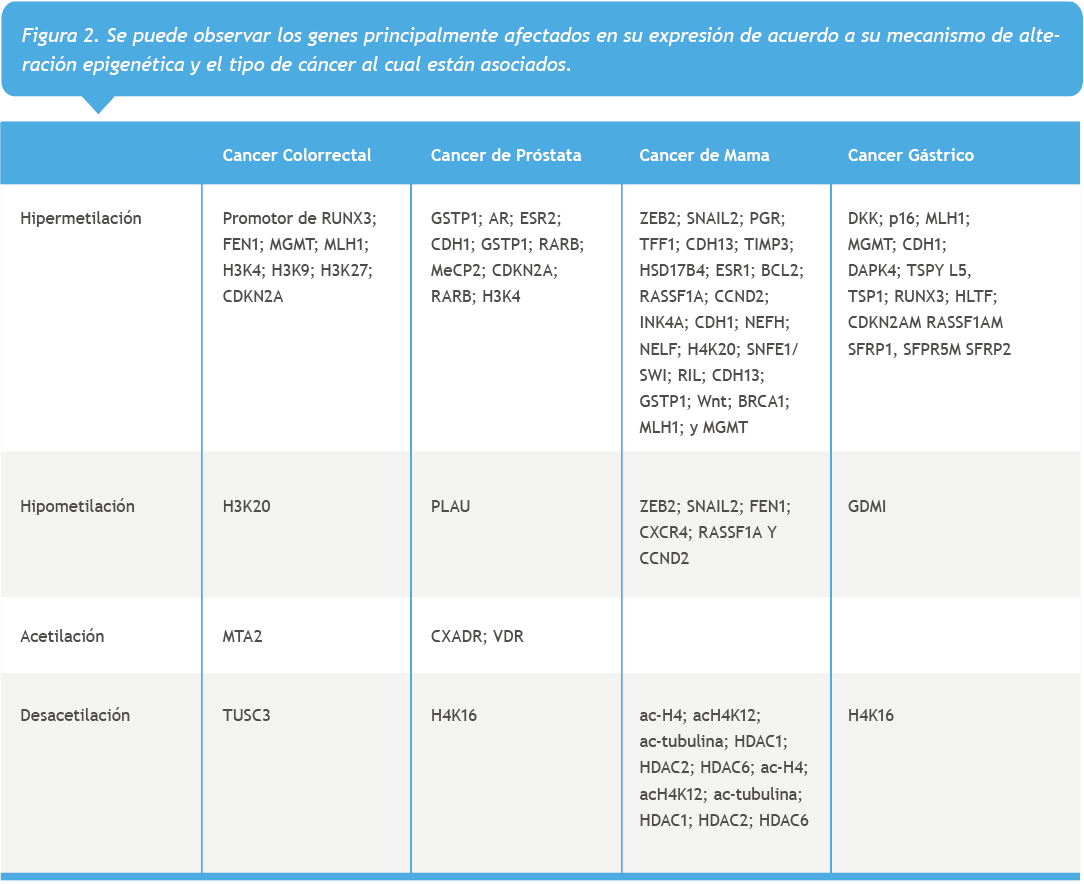
En el cáncer de próstata se encontraron cambios de metilación e hipermetilación de los genes: GST-1, AR, ER, CDKN2A y CDH1 así como hipometilación relacionada con los genes DNMT1 y PLAU. En el cancer de mama se encontraron cambios epigenéticos asociados a hipermetilación en genes que influyen en la reparación del DNA (BRCA1, MLH1 y MGMT), así como en el gen CDH1, de igual manera se ha encontrado relación entre la metilación de RASSF1A, CCND2, GSTP1, TWIST , PGR, TFF1, CDH13, ZEB2 y SNAIL2.
En la neoplasia colorrectal se han asociado cambios epigeneticos con su desarrollo consistentes en metilación de genes que actúan en vías de señalización como la Wnt con los genes APC y AXIN2, así como de otros cuya función radica en vías de reparación del DNA incluyendo entre estos genes los MLH1, MLH2, CDKN2A, y MGMT. Cabe agregar que se ha encontrado patrones distintivos en la metilación de islas CpG, lo cual se ha denominado Fenotipo de Metilación de Islas CpG o CIMP y cambios relacionados con hipoacetilación global en las Lisinas de histonas H3, H4, el gen MTA2, trimetilación de H3K20, concomitante a dimetilación y trimetilación de H3K4, H3K9 y H3K27.
También se han encontrado cambios relacionados con ncRNA en CRC y cáncer gástrico los cuales han demostrado utilidad como biomarcadores para el diagnóstico, gravedad y pronóstico dichas neoplasias. Por otra parte, en el cáncer gástrico se han encontrado cambios epigenéticos relacionados con supresiones de genes por medio de la metilación siendo estos: MLH1, P16, CDKN2A, CDH1, CDH4, DAPK, RAS, XIAS, XAF1, TSPY L5, SFRP2,SFRP1, SFPR5, SFRP2, HLTF, RUNX3 y TSP1.
En conclusión, dentro de las alteraciones epigenéticas relacionadas entre las cuatro neoplasias se observó que tienden a modificar la expresión de genes cuya implicados en vías de reparación de DNA y el control del ciclo celular. Los cambios epigenéticos más frecuentes incluyen la metilación y la hipermetilación, además cabe resaltar que también se han descrito la acetilación o hipometilación aunque en menor proporción en cáncer colorrectal, gástrico, mama, y de próstata. La expresión de la metilación de la región promotora de ciertos genes, propios de cada cáncer como por ejemplo BRCA1 en cáncer de mama guardan relación importante, también existen relaciones que pueden contribuir al desarrollo de cáncer como lo son metilación en las DNMT1 que están presentes tanto en cáncer de mama como en cáncer de colon, su hipermetilación conduce a la desmetilación de otras proteínas que conllevan a una desregulación del ciclo celular, mientras que su hipometilación es un proceso que se acumula con la edad del paciente, el número de divisiones y se asocia con el pobre pronóstico en el cáncer.
Entre las principales alteraciones epigenéticas asociadas con el desarrollo de cáncer de próstata, mama, colorrectal y gástrico se ha identificado con mayor prevalencia en cáncer gástrico la hipermetilacion del promotor de MLH1 hasta en el 71% de los casos, el daño generado en las vías de reparación celular se ha visto como manifestación tardía en el cáncer de cólon, y en cáncer de mama cuando a ésta se le suma una alteración de BRCA1, y MGMT, esta última se encuentra hipermetilada en cáncer gástrico y metilada en cáncer de colon.
La metilación en el promotor de CDKN2A se asocia a malignidad principalmente en cáncer de colon, aunque se ha dilucidado esta misma alteración en cáncer gástrico, la metilación específica en el exón 2 de esta región promotora se encuentra en más de la mitad de los cánceres de próstata mientras que la hipermetilación se encuentra en casos aislados. Es importante tener en cuenta que las alteraciones que conllevan al silenciamiento génico de la DNMT1 a través de mutaciones en la vía K-ras se ha visto relacionada tanto en cáncer gástrico como en cáncer colorrectal. La hipermetilación de las islas CpG se han descrito alteraciones para ESR2 tanto en cáncer de próstata como en cáncer de mama, en este último se asocia con alteraciones de BRCA1, y las vías de señalización de Wnt con los genes APC y AXIN2. Uno de los factores implicados que se asocia a malignidad en cáncer de mama, y gástrico es la hipermetilación de CDH1 en cáncer de mama conlleva a un peor pronóstico ya que se potencia la posibilidad de metástasis, en cuanto al cáncer gástrico estas alteraciones se relacionan con infección por H. pylori y se aumenta directamente proporcional con la progresión del tumor. El fenotipo de metilación de islas CpG en cáncer de cólon o su hipermetilación en cáncer gástrico contiene un alto porcentaje de metilación de promotores de genes de manera aberrante, existen varios subgrupos que se correlacionan con la presencia o ausencia de inestabilidad de microsatélites, mutaciones en BRAF, K- ras y p53, su hipermetilación ocurre en más de un 50% de los cánceres gástricos cuando se hipermetilan las regiones CpG de TSPY L5, la hipermetilación de GSTP1 se vio implicada en cáncer de próstata.
Por último se debe tener en cuenta que la hipoacetilación global de las lisinas de H3 y H4 se ha visto implicada en estadios más tardíos en cáncer de colon y se relaciona con escasa regulación de genes supresores de tumores.
Teniendo en cuenta que el conocimiento de estas alteraciones repercuten las propuestas de blancos terapéuticos es posible observar que las neoplasias estudiadas tiene una gran variedad de cambios epigenéticos asociados, coexistiendo varias de estas alteraciones al mismo tiempo, por lo cual el desarrollo de técnicas más avanzadas para identificación rápida de cambios epigenéticos asociados a neoplasias es necesario para el avance médico en el diagnóstico las neoplasias estudiadas, ya que las alteraciones revisadas tienen utilidad como biomarcadores para determinar el diagnóstico, la susceptibilidad, gravedad y pronóstico de estas patologías tan prevalentes en nuestra región. Por tanto, cabe decir que la medicina debe encaminarse hacia la creación y mejoramiento de tecnologías que permitan prevenir estos cambios epigenéticos y promover un manejo más profundo y completo a estas patologías.
Como muestra de los avances a nivel diagnóstico, se encontró que se ha investigado la presencia de biomarcadores de metilación de ADN en heces y sangre.67 De igual manera la determinación del estado de metilación de NPY, PENK y WIF1 como medida de tamizaje para cáncer colorrectal, y así identificar individuos a los cuales se les debería hacer colonoscopia.68 En cáncer de próstata la FDA aprobó en el 2016 la prueba PROGENSA PCA3, una prueba específica de próstata que mediante la toma de una muestra de orina permite detectar la presencia de lncRNA PCA3, las cuales se han asociado a la supervivencia de células tumorales, también teniendo función como parámetro para determinar la necesidad de repetir biopsias en caso de que esta sea negativa y la prueba de orina positiva.69
El estadio clínico del cáncer de mama se ha correlacionado con la metilación y acetilación la cual permite diferenciar entre células benignas y cancerosas. Estos patrones epigenéticos en las histonas podrían representar un signo temprano en el cáncer de mama.
Se ha encontrado que niveles altos de acetilación y metilación obedecen a un pronóstico más favorable en el cáncer de mama tipo luminal mientras que los niveles más bajos de metilación y acetilación de histonas en carcinoma basal triple negativo y los subtipos HER2 se asociaron a peor pronóstico. Otras investigaciones mostraron que la metilación del gen GHSR permite con una alta sensibilidad y especificidad diferenciar el tejido mamario normal o benigno del carcinoma ductal invasivo.41
Recomendaciones
Dada la importancia de encontrar tratamientos menos invasivos, se recomienda realizar un análisis a las principales dianas terapéuticas en los cánceres revisados. De igual manera, teniendo en cuenta la urgencia por encontrar marcadores tumorales que nos sirvan como métodos diagnósticos más sensibles y específicos, se recomienda investigar los cambios en la expresión de moléculas de acuerdo con las etapas de cada uno de los cánceres vistos. Se sugiere incluir en próximos artículos un apartado sobre la diferencia en progresión y sobrevida de la enfermedad de acuerdo con las variaciones epigenéticas que tenga el tipo de cáncer.
Abreviaturas
APC: Adenomatous polyposis coli/Poliposis coli adenomatosa; AR: Androgen receptor/Receptor de andrógenos; ATM: ATM serine/threonine kinase /ATM serina/treonina quinasa; AXIN2: Axis Inhibition Protein 2/Proteína de inhibición del eje 2; BCL2: BCL2 apoptosis regulator/Regulador de apoptosis BCL2; BRAF: B-Raf proto-oncogene, serine/threonine kinase / Protooncogen BRaf, serina/treonina quinasa; BRCA1: Breast Cancer Type 1 Susceptibility Protein/Proteína de susceptibilidad al cáncer de mama tipo 1; CBP: CREB binding protein/Proteína de unión a CREB; CCND2: Cyclin D2/Ciclina D2; CDH1: Cadherin 1 or cadherin E/Cadherina 1 o cadherina E; CDH13: Cadherin 13/Cadherina 13; CDKI: Cyclin-dependent Kinase Inhibitor/Inhibidor de quinasa dependiente de ciclina; CDKN2A/P16/P14/INK4A: Cyclindependent Kinase Inhibitor 2A/Inhibidor de quinasa dependiente de ciclina 2A; CDKN2B/P15: Cyclin-dependent Kinase Inhibitor 2B/inhibidor de quinasa dependiente de ciclina 2B; CIMP: CpG island methylator phenotype /Fenotipo metilador de islas CpG; CRC: Colorectal cancer/Cáncer colorectal; CXADR: CXADR Ig-like cell adhesion molecule/Molécula de adhesión celular tipo CXADR Ig; CXCL12: C-X-C motif chemokine ligand 12/Motivo CXC quimiocina ligando 12; CXCR4: C-X-C motif chemokine receptor 4/Receptor de quimiocina con motivo CXC 4; DAPK: Death associated protein kinase/Proteína quinasa asociada a muerte; DKK: Dickkopf-related protein/Proteína relacionada con dickkopf; DNMT 1: DNA methyltransferase 1/ Metiltransferasa de DNA tipo 1; ER: Estrogen receptor/Receptor de estrógeno; ESR: Estrogen receptor/Receptor de estrógeno; EZH2: Enhancer of zeste 2 polycomb repressive complex 2 subunit/potenciador de la subunidad 2 del complejo represivo polycomb zeste 2; FEN1: Flap structure-specific endonuclease 1 /Endonucleasa FLAP 1; GSTP1: Glutathione S-transferase pi 1/Glutatión S-transferasa pi 1; HAT: Histone Acetyltransferase/Acetiltransferasa de histona; HDAC: Histone Deacetylase/Histona desacetilasa; HER2: erb-b2 receptor tyrosine kinase 2/Receptor de erb-b2 tirosina quinasa 2; HIN1: Secretoglobin family 3A member 1/Miembro 1 de la familia 3A de secretoglobina; HLTF: Helicase like transcription factor/Factor de transcripción parecido a la helicasa; HOTAIR: HOX transcript antisense RNA/ARN antisentido de la transcripción de HOX; HSD17B4: Hydroxysteroid 17-beta dehydrogenase 4/Hidroxiesteroide 17-beta deshidrogenasa 4; HSRBC: Caveolae associated protein 3/Proteína 3 asociada a caveolae; HTM: Histone methyltransferase/Metiltransferasa de histonas; IARC: International Agency for research on Cancer/Centro Internacional de Investigaciones sobre el Cáncer; KAT7: Lysine acetyltransferase 7/Lisina acetiltransferasa 7; KDM: Lysine demethylases/ Demetilasa de lisina; KLF2: Kruppel like factor 2/Factor similar a Kruppel 2; K-ras: KRAS proto-oncogene, GTPase/Protooncogen KRAS, GTPasa; LNCaP: Androgen-sensitive human prostate adenocarcinoma cells/Células adenocarcinomatosas sensibles a andrógenos; lncRNA: Long non-coding RNA/ARN largos no codificantes; LSD1/KDM1A: Lysine demethylase 1A/Lisina desmetilasa 1A; MAPK: Mitogen-activated protein kinase/Proteína quinasa 1 activada por mitógeno; MBD1: Methyl-CpG binding domain protein 1/Proteína 1 de unión de dominio metil-CpG; MeCP2: methyl-CpG binding protein 2/Proteína de unión a metil-CpG 2; MGMT: O-6-methylguanine-DNA methyltransferase /O-6-metilguanina-ADN metiltransferasa; miRNA: micro-RNA/Micro ARN; MLH1, 2: mutL homolog 1/mutL homólogo 1; MSI: Microsatellite instability/Inestabilidad de microsatélites; MTA2: Metastasis associated 1 family member 2/Miembro de la familia 2 asociada a metástasis 1; MYC: MYC proto-oncogene/ Protooncogen MYC; ncRNA: non-coding RNA/RNA no codificante; NEFH: Neurofilament heavy/Neurofilamento pesado; NELF: Negative elongation factor/Factor de elongación negativo; NIH: National Institutes of Health/Instituto Nacional de Salud; NKD2: NKD inhibitor of WNT signaling pathway 2/Inhibidor 2 de NKD en la vía de señalización WNT; PI3K/AKT: signaling pathway phosphatidylinositol 3-kinase/serine-threonine protein kinase / Vía de señalización fosfatidilinositol 3 kinasa/proteína quinasa de serina-treonina; piRNAs: P-element-induced wimpy testis (PIWI)-interacting RNAs/Proteína PIWI de interacción RNA; PLAU: Urokinase-type plasminogen activator/Activador de plasminógeno de tipo uroquinasa; PR/PGR: Progesterone receptor/Receptor de progesterona; PRC1: Protein regulator of cytokinesis 1/Regulador de proteínas de la citocinesis 1; PRC2: Polycomb repressive complex 2/Complejo repressor polycomb 2; PSA: Prostate-specific antigen/Antígeno prostático específico; RARB: Retinoic acid receptor beta/Receptor de ácido retinoico beta; RASSF1A: Ras association domain family member 1/ Proteína de la familia de asociación ras 1; RB: Retinoblastoma; RIL: PDZ and LIM domain 4/PDZ y LIM dominio 4; RUNX3: RUNX family transcription factor 3/Factor de transcripción de la familia RUNX 3; SFRP2: Secreted frizzled-related protein 2/ Proteína secretada 2 relacionada con el frizz; SMRT: Nuclear receptor corepressor 2/Receptor nuclear corepressor 2; SNAI2: Snail family transcriptional repressor 2/Represor transcripcional de la familia SNAI2; SNFE1/SWI: SWI/SNF related/Regulador de cromatina relacionado con SWI/SNF; STAT3: Signal transducer and activator of transcription 3/Transductor de señal y activador de la transcripción 3; TERC: Telomerase RNA component/ Componente de ARN de telomerasa; TERT: Telomerase reverse transcriptase/Telomerasa transcriptasa inversa; TFF1: Trefoil factor 1/Factor trébol 1; TIMP3: TIMP metallopeptidase inhibitor 3/TIMP inhibidor de metalopeptidasa 3; TP53: Tumor protein P53/Proteína tumoral P53; TSP1: Thrombospondin 1 / Trombospondina 1; TSPY1: Testis specific protein Y-linked 1/ Proteína específica de testículo ligada a Y 1; TSPYL5: Testisspecific protein, Y-encoded-like 5/Proteína específica del testículo, codificada en Y como 5; TUSC3: Tumor Suppressor Candidate 3/Supresor tumoral candidato 3; TWIST1: Twist family bHLH transcription factor 1/Factor de transcripción bHLH de la familia twist 1; UHRF1: Ubiquitin like with PHD and ring finger domains 1/Ubiquitina como con PHD y dominios de dedo anular 1; VDR: Vitamin D receptor/Receptor de vitamina D; Wnt: Signaling pathway named “wingless e int-1” /Vía de señalización denominada “wingless e int-1”; XAF1: XIAP associated factor 1/Factor 1 asociado a XIAP; XIAP: X-linked inhibitor of apoptosis/Inhibidor de la apoptosis ligado al cromosoma X; ZEB2: Zinc finger E-box binding homeobox 2/Dedo de zinc E-box de unión homeobox 2; ZFAS1: ZNFX1 antisense RNA 1/ARN antisentido ZNFX1 1.
Agradecimientos
Este articulo es producto del proceso formativo de estudiantes de medicina de las asiganturas Biología del Cáncer y Escritura de artículos científicos biomédicos, ofrecidas por el Departamento de Ciencias Básicas de la Salud de la Pontificia Universidad Javeriana Cali. Los autores agradecen a Valentina Muñoz Chaves, diseñadora y productora de audio y video, por su apoyo en el diseño de los gráficos.
Referencias Bibliográficas
1. Iridoy-Zulet M, Pulido-Fontes L, Ayuso-Blanco T, Lacruz-Bescos F, Mendioroz-Iriarte M. Modificaciones epigenéticas en neurología: alteraciones en la metilación del ADN en la esclerosis múltiple. Neurología. 2017. 32:463-8.
2. Al-Aboud NM, Simpson B, Jialal I. Genetics, Epigenetic Mechanism. StatPearls Publishing: Florida; 2019 J. Disponible en: https://www.ncbi.nlm.nih.gov/books/NBK532999/
3. Shen H, Laird PW. Interplay between the cancer genome and epigenome. Cell. 2013; 153(1):38-55. DOI: 10.1016/j. cell.2013.03.008
4. Kondo Y. Epigenetic cross-talk between DNA methylation and histone modifications in human cancers. Yonsei Medical Journal. 2009. 50:455.63.
5. World Health Organization. Cancer Today: International Agency for Research on Cancer (IARC). Disponible en: https://gco.iarc.fr/ today/data/factsheets/cancers/10_8_9-Colorectum-fact-sheet.pdf.
6. World Health Organization. Cancer Today: International Agency for Research on Cancer (IARC) Colombia Factsheets. Disponible en:https://gco.iarc.fr/today/data/factsheets/populations/170-colombiafact- sheets.pdf.
7. Universidad del Valle. Registro Poblacional de Cancer de Cali (RPCC). Disponible en: http://rpcc.univalle.edu.co/es/index.php.
8. Epigenética. National Human Genome Research Institute. 2018. Disponible en: https://www.genome.gov/es/genetics-glossary/ Epigenetica
9. Biswas S, Rao CM. Epigenetic tools (The Writers, The Readers and The Erasers) and their implications in cancer therapy. Eur J Pharmacol. 2018; 837:8-24.
10. Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev. 2011; 25(10):1010-22.
11. García R, Ayala PA, Perdomo SP. Epigenética: definición, bases moleculares e implicaciones en la salud y en la evolución humana. Rev Cienc Salud. 2012; 10(1):59-71.
12. International Agency for Research on Cancer, Global Cancer Observatory. (2019). Cancer today. Disponible en: https://gco.iarc.fr/today/online-analysis-multi-bars?v=2018&mode=cancer&mode_ population=countries&population=900&populations=170&k ey=total&sex=0&cancer=39&type=0&statistic=5&prevalen ce=0&population_group=0&ages_group%5B%5D=0&ages_ group%5B%5D=17&nb_items=10&group_cancer=1&include_ nmsc=1&include_nmsc_other=1&type_multiple=%257B%2522 inc%2522%253Afalse%252C%2522mort%2522%253Atrue%25 2C%2522prev%2522%253Afalse%257D&orientation=horizont al&type_sort=0&type_nb_items=%257B%2522top%2522%253 Atrue%252C%2522bottom%2522%253Afalse%257D&populati on_group_globocan_id=#collapse-group-0-1
13. Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature. 1983; 301(5895):89-92.
14. Coppedè F. The role of epigenetics in colorectal cancer. Expert Rev Gastroenterol Hepatol. 2014; 8(8):935-48.
15. Khare S, Verma M. Epigenetics of colon cancer. Methods Mol Biol. 2012; 863:177-85.
16. Toyota M, Suzuki H, Yamamoto E, Yamano H, Imai K, Shinomura Y. Integrated analysis of genetic and epigenetic alterations in cancer. Epigenomics. 2009; 1(2):291-9.
17. Lotem J, Levanon D, Negreanu V, Bauer O, Hantisteanu S, Dicken J, et al. Runx3 at the interface of immunity, inflammation and cancer. Biochimica et Biophysica Acta (BBA). Reviews on Cancer. 2015; 1855(2):131-143.
18. Okugawa Y, Grady W. Epigenetic Alterations in Colorectal Cancer: Emerging Biomarkers. Gastroenterology. 2015; 149(5):1204-1225.
19. Nusse R. Wnt signaling in disease and in development. Cell Res. 2005; 15(1):28-32.
20. Taniue K, Hayashi T, Kamoshida Y, et al. UHRF1-KAT7-mediated regulation of TUSC3 expression via histone methylation/acetylation is critical for the proliferation of colon cancer cells. Oncogene. 2019. DOI: 10.1038/s41388-019-1032-y
21. Aravin AA, Sachidanandam R, Girard A, Fejes-Toth K, Hannon GJ. Developmentally regulated piRNA clusters implicate MILI in transposon control. Science. 2007; 316(5825):744-747. DOI: 10.1126/science.1142612
22. Jia L, Zhang S, Cheng B. Epigenetic roles of PIWI-interacting RNAs (piRNAs) in cancer metastasis (Review). Oncology Reports. 2018; 40(5):2423-2434. DOI: 10.3892/or.2018.6684
23. USCS Data Visualizations-CDC. Centers for Disease Control and Prevention. 2019. Disponible en: https://gis.cdc.gov/cancer/USCS/ DataViz.html
24. Bravo LE, Muñoz N. Epidemiology of cancer in Colombia. Colombia Médica. 2018; 49(1):09-12. DOI: 10.25100/cm.v49i1.3877
25. Chiam K, Ricciardelli C, Bianco-Miotto T. Epigenetic biomarkers in prostate cancer: Current and future uses. Cancer Letters. 2014; 342(2):248-256. DOI: 10.1016/j.canlet.2012.02.011
26. Lee W H, Isaacs WB, Steven-Bova G, Nelson WG. CG island methylation changes near the GSTP1 gene in prostatic carcinoma cells detected using the polymerase chain reaction: A new prostate cancer biomarker. Cancer Epidemiology Biomarkers and Prevention. 1997; 6(6):443-450.
27. Li LC, Carroll PR, Dahiya R. Epigenetic changes in prostate cancer: Implication for diagnosis and treatment. Journal of the National Cancer Institute. 2005; 97(2):103-115. DOI: 10.1093/jnci/dji010
28. Prins GS, Korach KS. The role of estrogens and estrogen receptors in normal prostate growth and disease. Steroids. 2008; 73: 233-244. DOI: 10.1016/j.steroids.2007.10.013
29. Verdoodt B, Sommerer F, Palisaar RJ, Noldus J, Vogt M, Nambiar S, Neid M. Inverse association of p16 INK4a and p14 ARF methylation of the CDKN2a locus in different Gleason scores of prostate cancer. Prostate Cancer and Prostatic Diseases. 2011; 14(4):295-301. DOI: 10.1038/pcan.2011.45
30. Gravdal K, Halvorsen OJ, Haukaas SA, Akslen LA. A switch from E-cadherin to N-cadherin expression indicates epithelial to mesenchymal transition and is of strong and independent importance for the progress of prostate cancer. Clin Cancer Res. 2007; 13(23):7003-11.
31. Yegnasubramanian S. Prostate cancer epigenetics and its clinical implications. Asian Journal of Andrology. 2016; 18(4):549-558. DOI: 10.4103/1008-682X.179859
32. Andrés G, Ashour N, Sánchez-Chapado M, Ropero S, Angulo J C. The study of DNA methylation in urological cancer: Present and future. Actas Urologicas Espanolas. 2013; 37(6):368-375. DOI: 10.1016/j.acuro.2013.03.001
33. Jerónimo C, Bastian PJ, Bjartell A, Carbone GM, Catto JWF, Clark SJ, Shariat SF. Epigenetics in prostate cancer: Biologic and clinical relevance. European Urology. 2011; 60(4):753-766. DOI: 10.1016/j.eururo.2011.06.035
34. Campbell MJ, Trump DL. Vitamin D Receptor Signaling and Cancer. Endocrinology and Metabolism Clinics of North America. 2017; 46(4):1009-1038. DOI: 10.1016/j.ecl.2017.07.007
35. Duffy MJ. Biomarkers for prostate cancer: prostate-specific antigen and beyond. Clinical Chemistry and Laboratory Medicine (CCLM). 2019; 0(0). DOI: 10.1515/cclm-2019-0693
36. Frame FM, Maitland NJ. Epigenetic control of gene expression in the normal and malignant human prostate: A rapid response which promotes therapeutic resistance. International Journal of Molecular Sciences. 2019; 20(10):31. DOI: 10.3390/ijms20102437
37. Coleman M, Quaresma M, Berrino F, Lutz J, De Angelis R, Capocaccia R, et al. Cancer survival in five continents: a worldwide population-based study (CONCORD). The Lancet Oncology. 2008; 9(8):730-756.
38. Organización Mundial de la Salud. Cáncer de mama. Prevención y control. WHO. World Health Organization; 2015. Disponible en: http://www.who.int/topics/cancer/breastcancer/es/index3.html
39. Cáncer de mama: Estadísticas. American Society of Clinical Oncology. 2019. Disponible en: https://www.cancer.net/es/tiposde- cáncer/cáncer-de-mama/estadísticas
40. Li CI, Uribe DJ, Daling JR. Clinical characteristics of different histologic types of breast cancer. Br J Cancer. 2005; 93(9):1046-52.
41. Byler S, Goldgar S, Heerboth S, Leary M, Housman G, Moulton K, et al. Genetic and epigenetic aspects of breast cancer progression and therapy. Anticancer Res. 2014; 34(3):1071-7.
42. Abdel-Hafiz HA, Horwitz KB. Role of epigenetic modifications in luminal breast cancer. Epigenomics. 2015; 7(5):847-62.
43. Karsli-Ceppioglu S, Dagdemir A, Judes G, Ngollo M, Penault- Llorca F, Pajon A, et al. Epigenetic mechanisms of breast cancer: An update of the current knowledge. Epigenomics. 2014; 6(6):651-64.
44. Wu Y, Sarkissyan M, Vadgama J V. Epigenetics in Breast and Prostate Cancer. In: Verma M (editor). Cancer Epigenetics: Risk Assessment, Diagnosis, Treatment, and Prognosis. Springer: New York; 2015. p. 425-66. DOI: 10.1007/978-1-4939-1804-1_23
45. Singh AK, Pandey A, Tewari M, Shukla HS, Pandey HP. Epigenetic silencing of BRCA1 gene associated with demographic and pathologic factors in sporadic breast cancer: A study of an Indian population. Eur J Cancer Prev. 2011; 20(6):478-83.
46. Aristizábal-Pachón AF, Satie-Takahashi C. Efecto de las alteraciones genéticas y epigenéticas de la cadherina E y su expresión en la transcripción en la propensión al cáncer de mama. Biomedica. 2016; 36(4):593-602.
47. Calmon MF, Jeschke J, Zhang W, Dhir M, Siebenkäs C, Herrera A, et al. Epigenetic silencing of neurofilament genes promotes an aggressive phenotype in breast cancer. Epigenetics. 2015 Jan 1; 10(7):622-32.
48. Li L, Lee KM, Han W, Choi JY, Lee JY, Kang GH, et al. Estrogen and progesterone receptor status affect genome-wide DNA methylation profile in breast cancer. Hum Mol Genet. 2010; 19(21):4273-7.
49. Mehrotra J, Ganpat MM, Kanaan Y, Fackler MJ, McVeigh M, Lahti-Domenici J, et al. Estrogen Receptor/Progesterone Receptor- Negative Breast Cancers of Young African-American Women Have a Higher Frequency of Methylation of Multiple Genes than Those of Caucasian Women. Clin Cancer Res. 2004; 10(6):2052-7.
50. Ramos EAS, Grochoski M, Braun-Prado K, Seniski GG, Cavalli IJ, Ribeiro EMSF, et al. Epigenetic changes of CXCR4 and its ligand CXCL12 as prognostic factors for sporadic breast cancer. PLoS One. 2011; 6(12).
51. Singh P, Yang M, Dai H, Yu D, Huang Q, Tan W, et al. Overexpression and hypomethylation of Flap endonuclease 1 gene in breast and other cancers. Mol Cancer Res. 2008; 6(11):1710-7.
52. Vrba L, Muñoz-Rodríguez JL, Stampfer MR, Futscher BW. miRNA Gene Promoters Are Frequent Targets of Aberrant DNA Methylation in Human Breast Cancer. PLoS One. 2013; 8(1):e54398. DOI: 10.1371/journal.pone.0054398
53. Elsheikh SE, Green AR, Rakha EA, Powe DG, Ahmed RA, Collins HM, et al. Global histone modifications in breast cancer correlate with tumor phenotypes, prognostic factors, and patient outcome. Cancer Res. 2009; 69(9):3802-9.
54. Situación de cáncer en Colombia. Cuenta de Alto Costo. Fondo Colombiano de Enfermedades de Alto Costo. Capitulo 12. 2015 Disponible en: https://cuentadealtocosto.org/site/images/Libro%20 Cáncer%202015/18%20-%20Cap%C3%ADtulo%2012%20-%20Cáncer%20Gástrico.pdf.
55. Pardo-Ramos C. Atlas de mortalidad por cáncer en Colombia. Cuarta edición. Instituto Nacional de Cancerología ESE: Bogotá; 2017.
56. Calcagno DQ, Gigek CO, Chen ES, Burbano RR, Smith M de AC. DNA and histone methylation in gastric carcinogenesis. World J Gastroenterol. 2013; 19(8):1182-92.
57. Fu DG. Epigenetic alterations in gastric cancer (review). Mol Med Rep. 2015; 12(3):3223-30.
58. Graziano F, Arduini F, Ruzzo A, Bearzi I, Humar B, More H, et al. Prognostic Analysis of E-Cadherin Gene Promoter Hypermethylation in Patients with Surgically Resected, Node-Positive, Diffuse Gastric Cancer. Clin Cancer Res. 2004; 10(8):2784-9.
59. Leodolter A, Alonso S, González B, Ebert MP, Vieth M, Röcken C, et al. Somatic DNA hypomethylation in H. Pylori-Associated highrisk gastritis and gastric cancer: Enhanced somatic hypomethylation associates with advanced stage cancer. Clin Transl Gastroenterol. 2015; 6(4):85.
60. Pirini F, Noazin S, Jahuira-Arias MH, Rodriguez-Torres S, Friess L, Michailidi C, et al. Early detection of gastric cancer using global, genome-wide and IRF4, ELMO1, CLIP4 and MSC DNA methylation in endoscopic biopsies. Oncotarget. 2017; 8(24):38501-16.
61. Nie F, Yu X, Huang M, Wang Y, Xie M, Ma H, et al. Long noncoding RNA ZFAS1 promotes gastric cancer cells proliferation by epigenetically repressing KLF2 and NKD2 expression. Oncotarget. 2017; 8(24):38227-38.
62. Lü MH, Tang B, Zeng S, Hu CJ, Xie R, Wu YY, et al. Long noncoding RNA BC032469, a novel competing endogenous RNA, upregulates hTERT expression by sponging MIR-1207-5p and promotes proliferation in gastric cancer. Oncogene. 2016; 35(27):3524-34.
63. Chen WM, Huang M De, Sun DP, Kong R, Xu TP, Xia R, et al. Long intergenic non-coding RNA 00152 promotes tumor cell cycle progression by binding to EZH2 and repressing p15 and p21 in gastric cancer. Oncotarget. 2016; 7(9):9773-87.
64. Liu X hua, Sun M, Nie F qi, Ge Y bin, Zhang E bao, Yin D dan, et al. Lnc RNA HOTAIR functions as a competing endogenous RNA to regulate HER2 expression by sponging miR-331-3p in gastric cancer. Mol Cancer. 2014; 13(1):92.
65. Bouhaddou M, Yu LJ, Lunardi S, Stamatelos SK, Mack F, Gallo JM, et al. Predicting In Vivo Efficacy from In Vitro Data: Quantitative Systems Pharmacology Modeling for an Epigenetic Modifier Drug in Cancer. Clin Transl Sci. 2019. DOI: 10.1111/cts.12727
66. Haftchenary S, Avadisian M, Gunning PT. Inhibiting aberrant Stat3 function with molecular therapeutics: A progress report. Anticancer Drugs. 2011; 22(2):115-27.
67. Javier-Carmona F, Azuara D, Berenguer-Llergo A, Fernández AF, Biondo S, De Oca J, et al. DNA methylation biomarkers for noninvasive diagnosis of colorectal cancer. Cancer Prev Res. 2013; 6(7):656-65.
68. Roperch JP, Incitti R, Forbin S, Bard F, Mansour H, Mesli F, et al. Aberrant methylation of NPY, PENK, and WIF1 as a promising marker for blood-based diagnosis of colorectal cancer. BMC Cancer. 2013; 13.
69. Lemos AEG, Ferreira LB, Batoreu NM, Freitas PPD, Bonamino MH, et al. PCA3 long noncoding RNA modulates the expression of key cancer-related genes in LNCaP prostate cancer cells. Tumor Biology. 2016; 37(8):11339-48. DOI: 10.1007/s13277-016-5012-3

Más notas de la edición 128

Lee desde Issuu nuestra última edición publicada en Julio 2024, Edición número 155


Notas relacionadas a Descripción de las principales alteraciones...
